Le corticosurrénalome (ou carcinome corticosurrénalien) est une tumeur rare de la surrénale. Nous vous proposons, dans ce dossier, un aperçu de la stratégie thérapeutique ainsi que des innovations en cours de développement.
Résumé
Le corticosurrénalome est responsable d’un syndrome sécrétoire (hypercortisolisme, hyperandrogénie ou plus rarement hyperaldostéronisme) dans environ deux tiers des cas. La biopsie est en principe contre-indiquée, sauf situations très spécifiques discutées en RCP. La prise en charge doit être réalisée en centre expert (réseau ENDOCAN-COMETE) avec, pour objectif, une exérèse R0 de la tumeur dans les formes localisées. Le traitement adjuvant par mitotane ± chimiothérapie par EDP (étoposide-doxorubicine-cisplatine) dépend du niveau de risque. Dans les formes avancées ou en rechute, le mitotane reste la pierre angulaire du traitement, généralement combiné avec des traitements locaux et/ou systémiques selon la charge tumorale et l’évolutivité. La chimiothérapie EDP, associée au mitotane, constitue le standard de première ligne en situation métastatique agressive.
Abstract
Adrenocortical carcinoma: standards of care and future perspectives
Adrenocortical carcinoma is a rare and aggressive malignancy arising from the adrenal cortex, associated with hormone hypersecretion (hypercortisolism, hyperandrogenism, or more rarely hyperaldosteronism) in approximately two thirds of cases. Percutaneous biopsy is generally contraindicated and should only be considered in very selected situations after discussion within an expert multidisciplinary tumor board. Management should be centralized in expert centers (ENDOCAN-COMETE network in France), with the primary objective of achieving complete (R0) surgical resection in localized disease. Adjuvant treatment with mitotane, with or without EDP chemotherapy, is guided by the estimated risk of recurrence. In advanced or relapsed disease, mitotane remains the cornerstone of systemic treatment, usually combined with locoregional and/or systemic therapies according to tumor burden and disease kinetics. In patients with aggressive metastatic disease, EDP chemotherapy (etoposide, doxorubicin, and cisplatin) in combination with mitotane is the standard first-line treatment.
Keywords: Adrenocortical carcinoma, Rare cancer, Hormone hypersecretion, Mitotane, EDP
Introduction
Les tumeurs surrénaliennes
Les tumeurs surrénaliennes sont des découvertes de plus en plus fréquentes dans la pratique clinique, généralement de façon fortuite à l’occasion d’un scanner abdomino-pelvien réalisé pour une autre indication. Si la majorité d’entre elles sont bénignes, non sécrétantes et sans incidence clinique, un petit pourcentage correspond à des tumeurs malignes. Chez l’adulte, les principales tumeurs primitives malignes de la surrénale sont les phéochromocytomes et les corticosurrénalomes.
Le corticosurrénalome
Le corticosurrénalome (ou carcinome corticosurrénalien) est une tumeur développée aux dépens du cortex surrénalien. Son incidence est estimée entre 0,5 et 2 cas par million d’habitants et par an, avec une légère prédominance féminine. Le pic d’incidence survient entre 40 et 60 ans, mais des formes pédiatriques existent, notamment dans des contextes de prédispositions génétiques comme les mutations de TP53 (syndrome de Li-Fraumeni) (1).
Une prise en charge spécialisée en centre expert
Le corticosurrénalome est une maladie rare, de mauvais pronostic, qui nécessite une prise en charge spécialisée en centre expert. En France, le réseau national de référence ENDOCAN-COMETE coordonne la prise en charge des cancers de la surrénale (phéochromocytomes et paragangliomes métastatiques ainsi que corticosurrénalomes), avec une relecture anatomo-pathologique spécialisée, des réunions de concertations pluridisciplinaires (RCP) dédiées au niveau régional et national, ainsi que des projets de recherche clinique et translationnelle (2). La liste des centres experts est disponible sur le site de la Société française d’endocrinologie : www.sfendocrino.org/les-centres-du-reseau-comete-cancers-de-la-surrenale/. Au niveau européen, les réseaux ENS@T (European Network for the Study of Adrenal Tumours) et EURACAN (European Reference Network for Rare Adult Solid Tumours) coordonnent les efforts de recherche et établissent des recommandations de prise en charge (3, 4).
Diagnostic
Le diagnostic de corticosurrénalome repose sur l’analyse histologique de la pièce opératoire ainsi que sur le bilan d’extension. Il n’existe cependant aucun marqueur histologique formel pour déterminer le caractère malin : seule la présence de métastases permet d’affirmer la malignité.
Il est important de noter que les biopsies sont généralement contre-indiquées en cas de masse surrénalienne suspecte de lésion maligne primitive, du fait du risque de dissémination et surtout de poussée hypertensive.
Signes cliniques évocateurs
Dans environ 60 à 70 % des cas, le corticosurrénalome est fonctionnel, c’est-à-dire responsable d’une production autonome d’hormones stéroïdiennes. Le tableau clinique dépend alors de la nature de la sécrétion :
• sécrétion de cortisol : syndrome de Cushing (prise de poids abdominal, HTA, diabète, fragilité cutanée, fonte musculaire, ostéoporose) ;
• sécrétion d’androgènes : signes de virilisation (hirsutisme, alopécie, troubles du cycle) ;
• sécrétion d’aldostérone (plus rare) : HTA résistante et hypokaliémie.
Les formes non fonctionnelles (30 à 40 %) peuvent être découvertes fortuitement ou sur des symptômes liés à l’effet de masse (douleur lombaire, compression vasculaire).
Bilan hormonal
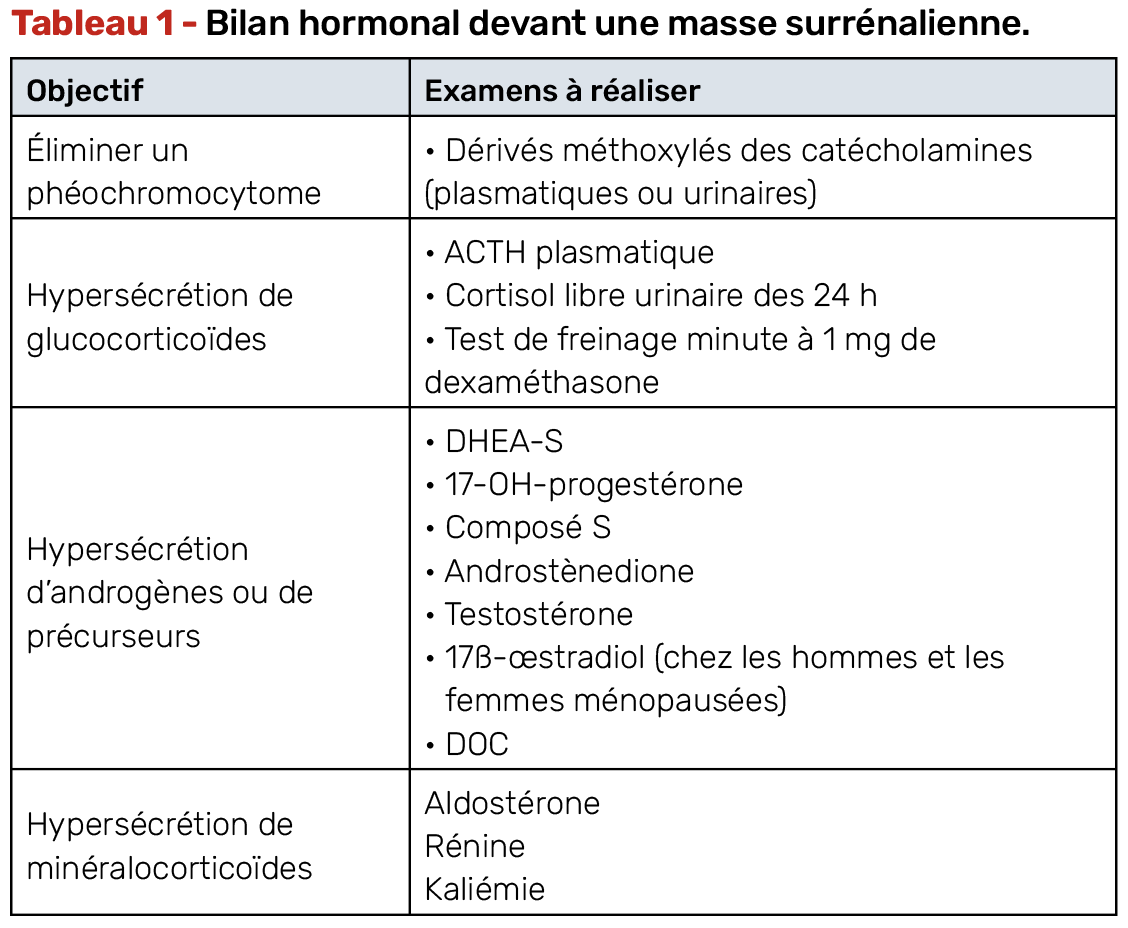
Tout patient porteur d’une masse surrénalienne, même asymptomatique, doit bénéficier d’un bilan hormonal complet (Tab. 1). Ce bilan vise à :
• éliminer un phéochromocytome, dont la prise en charge chirurgicale et surtout anesthésique est spécifique.
• dépister une sécrétion hormonale occulte (notamment un hypercortisolisme).
Ce bilan doit être réalisé avant toute intervention chirurgicale afin d’anticiper les complications liées au syndrome sécrétoire (déséquilibre glycémique, poussée hypertensive, etc.) et d’orienter le diagnostic étiologique.
Imagerie
L’imagerie est indispensable pour caractériser la lésion, évaluer son extension locorégionale et rechercher des métastases à distance.
• Le scanner thoraco-abdomino-pelvien sans et avec injection (au moins triphasique) est le premier examen à réaliser. Le corticosurrénalome est typiquement une masse > 6 cm, avec une densité spontanée > 10 UH, irrégulière, hétérogène, avec des zones nécrotiques, un rehaussement modéré et un wash-out incomplet (généralement inférieur à 50 % à 10 minutes).
• L’IRM surrénalienne n’est pas systématique, mais peut aider en cas de contre-indication au produit de contraste iodé.
• La TEP-FDG pré-opératoire est nécessaire en cas de suspicion de malignité (en faveur de la malignité en cas d’hyperfixation intense avec un rapport surrénale/foie > 1,45).
Contrairement aux phéochromocytomes, les autres traceurs de médecine nucléaire ont peu de place.
Anatomopathologie
La biopsie percutanée n’est pas recommandée en première intention, du fait d’un risque de dissémination tumorale et de relargage hormonal brutal en cas de forme sécrétante. Par ailleurs, elle ne permet pas toujours de trancher entre un adénome et un corticosurrénalome.
La biopsie est indiquée, après discussion en RCP experte, en cas de :
• lésion non résécable avec indication de traitement systémique ;
• suspicion de cancer extra-surrénalien (sarcome rétropéritonéal, lymphome, métastase d’un cancer solide).
Les marqueurs immunohistochimiques
Le diagnostic de certitude dépendra de l’analyse anatomopathologique de la pièce opératoire. La confirmation de l’origine corticosurrénalienne repose sur des marqueurs immunohistochimiques (dont le plus spécifique est SF1+). Le principal diagnostic différentiel au niveau surrénalien est le phéochromocytome qui peut exprimer certains marqueurs communs tels que la synaptophysine mais qui, contrairement au corticosurrénalome, exprime la chromogranine A.
Les scores histopronostiques
Il n’existe aucun marqueur histologique formel pour déterminer la malignité des corticosurrénalomes : seule la présence de métastase permet d’affirmer la malignité. Il existe cependant des scores histopronostiques, tels que le score de Weiss, qui permettent d’évaluer le potentiel de malignité. On estime ainsi qu’une tumeur corticosurrénalienne avec un score de Weiss ≥ 3 est à potentiel métastatique et donc à considérer comme un corticosurrénalome. Par ailleurs, parmi les corticosurrénalomes avérés, l’index de prolifération Ki67 est un facteur pronostique majeur qui va guider le choix du traitement adjuvant.
Oncogénétique et biologie moléculaire
Le corticosurrénalome est sporadique dans la plupart des cas, mais il existe des formes liées à des syndromes de prédisposition génétique, en lien avec des mutations de TP53, MEN1, APC, FH, SDHx, PRKAR1A. Une consultation de génétique est recommandée en cas d’âge jeune (< 40 ans), d’antécédents personnels ou familiaux, et d’atteinte bilatérale ou de phénotype syndromique.
Sur le plan somatique, le profil moléculaire des corticosurrénalomes est marqué par des altérations de la voie WNT-β-caténine, du cycle cellulaire et de l’apoptose (p53) (5). L’inclusion dans des programmes tels que le Plan France médecine génomique doit être encouragée afin de déterminer de nouvelles cibles thérapeutiques.
Prise en charge des corticosurrénalomes localisés
La prise en charge des corticosurrénalomes localisés repose sur la chirurgie, avec, dans certains cas, la nécessité d’un traitement adjuvant.
Préparation pré-opératoire
L’objectif est de réduire le risque de complications liées à la chirurgie et à l’hypercortisolisme éventuel. La coordination entre chirurgien, anesthésiste, endocrinologue et oncologue est cruciale.
• En cas de sécrétion de cortisol, une préparation par blocage stéroïdien (métopirone, kétoconazole, osilodrostat) peut être discutée.
• Un traitement substitutif par hydrocortisone devra dans tous les cas être instauré en péri-opératoire pour éviter une insuffisance surrénalienne aiguë (inhibition de l’axe hypothalamo-hypophysaire).
Chimiothérapie néoadjuvante
En cas de résécabilité incertaine, une chimiothérapie néoadjuvante peut être proposée. Elle consiste en deux à quatre cycles de chimiothérapie à base de platine (cisplatine étoposide ± doxorubicine). La chirurgie sera :
• réalisée en cas de réponse,
• rediscutée en cas de stabilisation,
• reportée en cas de progression.
Chirurgie
La chirurgie curative est le traitement de référence du corticosurrénalome localisé, avec pour objectif une résection complète sans effraction capsulaire.
• La voie chirurgicale ouverte par laparotomie est recommandée, en particulier si la tumeur est volumineuse (> 6 cm), suspecte d’envahissement local, ou si une surrénalectomie élargie est prévue.
• La cœlioscopie n’est envisageable que dans des centres experts, pour des tumeurs de petite taille et chez des patients sélectionnés.
• Le curage locorégional n’a pas fait la preuve de son effet sur la survie des patients, mais reste généralement pratiqué.
• La néphrectomie n’est pas systématique, mais peut être nécessaire dans le cadre de résections monobloc élargies.
Traitement adjuvant : mitotane ± chimiothérapie
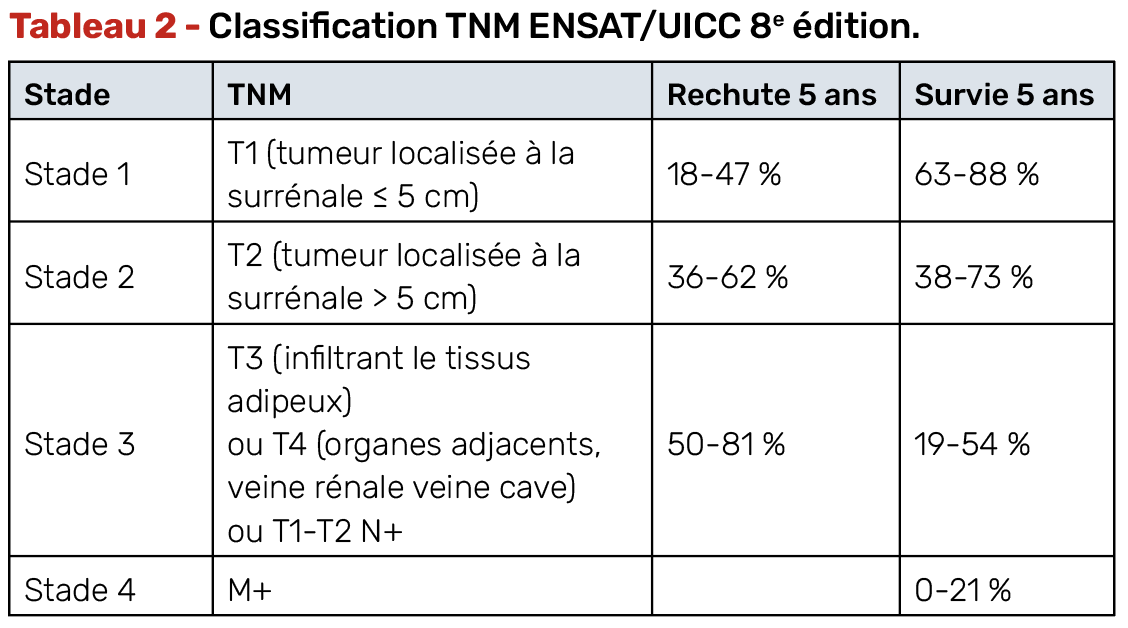
La décision d’un traitement adjuvant repose sur l’estimation du risque de récidive, fondée sur les critères suivants :
• stade ENSAT (I à IV) (Tab. 2),
• statut de résection (R0, R1, RX),
• index de prolifération Ki-67,
• présence de facteurs histopronostiques (nécrose, invasion vasculaire…).
La prise en charge adjuvante dépend directement de l’évaluation du risque de rechute (Tab. 3).

Mitotane adjuvant
Le mitotane (ou o,p’-DDD) est un dérivé de l’insecticide DDT, doté d’une action adrénolytique sélective sur le cortex surrénalien. C’est le seul médicament spécifiquement approuvé dans le traitement du corticosurrénalome. Il exerce une double action antisécrétoire (blocage des enzymes clés de la stéroïdogenèse) et antitumorale (effet cytotoxique par accumulation dans le tissu adréno-cortical).
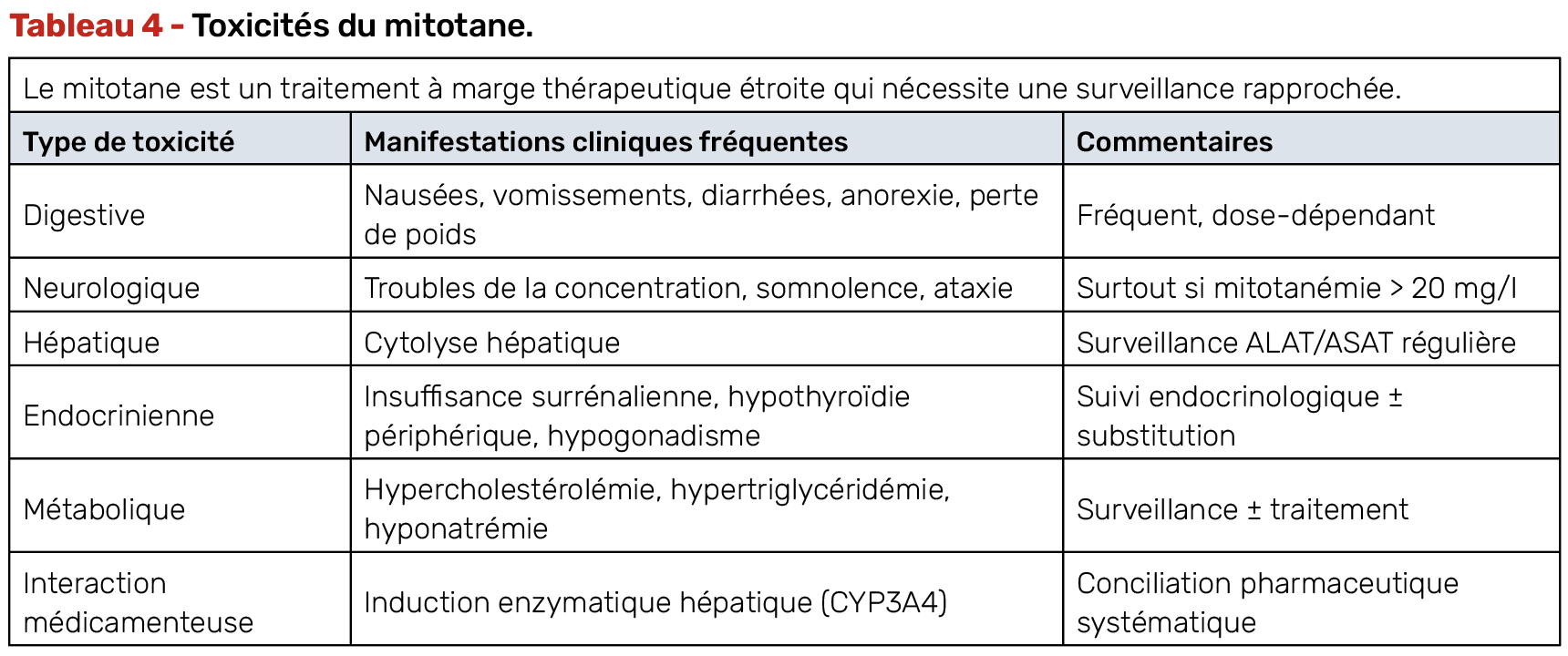
Il s’agit d’un médicament à marge thérapeutique étroite qui nécessite plusieurs mois pour être efficace. Il exige un suivi spécialisé, avec notamment des dosages réguliers pour aboutir à une concentration plasmatique entre 14 et 20 mg/l après une période d’instauration progressive. Il induit une insuffisance surrénalienne qui doit être substituée par hydrocortisone et expose à des toxicités multiples (Tab. 4).
Le traitement adjuvant par mitotane est recommandé chez les patients à haut risque de récidive (stade III, Ki-67 > 10 %, marges R1 ou RX) et discuté en situation de risque intermédiaire (stade II, Ki-67 5-10 %, marges R0). Il n’est en revanche plus recommandé chez les patients de risque faible, suite à l’étude ADIUVO qui avait comparé le mitotane pendant 24 mois par rapport à la surveillance chez 91 patients à faible risque de récidive (stade I-III R0, Ki-67 ≤10 %) et n’avait pas retrouvé de bénéfice en survie sans récidive, ni en survie globale (6).
Chimiothérapie adjuvante
Une étude rétrospective menée chez 31 patients suggère un bénéfice potentiel de la chimiothérapie adjuvante par EDP chez des patients à très haut risque (Ki-67 ≥ 30 %, stade III-IV, RX) (7). Cette stratégie est actuellement évaluée dans l’essai ADIUVO-2, qui randomise mitotane seul versus mitotane + EDP chez les patients à haut risque de rechute après chirurgie (clinicaltrials.gov/study/NCT03583710).
Radiothérapie adjuvante
En l’absence de données prospectives, la place de la radiothérapie adjuvante au niveau de la loge de surrénalectomie reste débattue. Elle est cependant recommandée en cas de chirurgie R1 ou Rx dans les stades III.
Suivi post-thérapeutique
Le suivi repose sur :
• une imagerie thoraco-abdomino-pelvienne tous les 3 à 6 mois pendant 2 ans, puis tous les 6 mois jusqu’à 5 ans, puis annuelle jusqu’à 10 ans ;
• un dosage des marqueurs hormonaux si la tumeur était sécrétante, pour détecter précocement une récidive.
Prise en charge des corticosurrénalomes avancés ou en rechute
Le pronostic des corticosurrénalomes avancés ou en rechute est globalement mauvais, avec une survie à 5 ans inférieure à 15 %. Cependant, il s’agit d’une maladie hétérogène avec parfois des formes indolentes ou de longs répondeurs aux traitements systémiques.
Rechutes
La prise en charge des rechutes après chirurgie dépend de l’intervalle libre.
• En cas d’intervalle libre prolongé (> 12 mois), les approches locorégionales, et notamment la chirurgie, sont discutées en première intention. Le mitotane est initié, s’il n’avait pas été prescrit en situation adjuvante.
• En cas d’intervalle libre court (< 6 mois), un traitement systémique est généralement indiqué.
Maladies avancées
Mitotane
Le traitement systémique des corticosurrénalomes métastatiques repose en premier lieu sur le mitotane. Du fait de sa latence pour atteindre un taux thérapeutique, il est généralement associé à des traitements locorégionaux (en cas de maladie oligométastatique et lentement évolutive) ou à la chimiothérapie (en cas de maladie agressive). L’interruption du mitotane se discute en cas d’intolérance et/ou de progression confirmée malgré une mitotanémie dans la zone thérapeutique de façon prolongée.
Traitements locorégionaux
Les traitements locorégionaux (chirurgie, radiothérapie, radiologie interventionnelle) sont utilisés en association au mitotane en cas d’atteinte oligométastatique. Ils permettent de réduire le volume tumoral et donc la sécrétion, et retardent le recours aux autres traitements systémiques.
Chimiothérapie : protocole EDP-mitotane
En cas de maladie plus agressive (Ki-67 > 20 %, nombreuses métastases, syndrome sécrétoire clinique), une chimiothérapie à base de platine sera ajoutée au mitotane. La chimiothérapie de référence est la combinaison EDP (étoposide 100 mg/m² J2-J3-J4, doxorubicine 40 mg/m² J1 et cisplatine 40 mg/m² J3-J4 toutes les 4 semaines) établie par l’essai randomisé FIRM-ACT (8). Cet essai randomisé comparait la combinaison EDP-mitotane à une bithérapie par streptozocine-mitotane chez 304 patients atteints de corticosurrénalomes avancés. Le critère de jugement principal de survie globale n’était pas atteint (médiane 14,8 versus 12,0 mois ; HR : 0,79 [IC 95 : 0,61-1,02]), mais le taux de réponse (23,2 versus 9,2 %) et la survie sans progression (médiane 5,0 versus 2,1 mois ; HR : 0,55 [IC 95 : 0,43-0,69]) étaient significativement améliorés. La tolérance du protocole EDP-mitotane est marquée par des toxicités fréquentes (asthénie, neutropénie, nausées). Le mitotane est initié 1 à 2 semaines avant la chimiothérapie, et poursuivi en entretien si efficace.
En cas d’échec du protocole EDP-mitotane
La prise en charge après échec d’EDP-mitotane repose sur des données limitées.
• Les TKI antiangiogéniques, tels que le cabozantinib, sont généralement utilisés en 2e ligne, hors AMM, sur la base d’études de phase II rapportant essentiellement des stabilisations (9).
• L’alternative est la reprise d’une chimiothérapie, avec, comme options, le témozolomide (avec un signal d’efficacité dans les tumeurs avec perte d’expression/méthylation du promoteur de MGMT), l’association gemcitabine + capécitabine ou encore la streptozocine (10, 11).
Immunothérapie
• Les traitements par anti-PD-(L)1 en monothérapie ont montré des taux de réponse de l’ordre de 20 % et ne disposent pas d’AMM (12).
• Plus récemment, un essai de phase II a évalué une association par camrélizumab (anti-PD1) + lapatinib (anti-VEGFR) et rapporté un taux de réponse prometteur de 52 % chez 21 patients atteints de corticosurrénalomes avancés pré-traités (13).
• D’autres approches d’immunothérapie ont été évaluées, notamment avec l’essai de phase I/II SPENCER qui évaluait le vaccin thérapeutique EO2401 (14). Le taux de réponse n’était que de 21 % dans la population globale, mais un sous-groupe semblait bénéficier à long terme du traitement, ce qui a motivé la poursuite de son développement.
Perspectives thérapeutiques
Parmi les pistes thérapeutiques, le ciblage de la voie WNT-β-caténine offre une perspective intéressante du fait de l’activation fréquente de celle-ci dans les corticosurrénalomes. Le CY-101, un peptide synthétique inhibiteur de la voie WNT-β-caténine, a montré un signal d’activité dans la phase I/IIa CICILIA, avec deux réponses durables parmi les six corticosurrénalomes traités, motivant une phase II attendue courant 2026.
Conclusion
Le corticosurrénalome est une maladie rare, agressive et complexe, dont la prise en charge nécessite une coordination étroite entre oncologue, endocrinologue et chirurgien, au sein d’un centre expert. La participation aux essais cliniques tels que ADIUVO-2 doit être encouragée pour faire progresser nos connaissances et donner accès aux meilleurs traitements.
Si les progrès thérapeutiques restent modestes jusqu’à présent, les développements récents autour des combinaisons immunothérapie/anti-angiogénique ou du ciblage de la voie WNT-β-caténine ouvrent de nouvelles perspectives.

L’auteur déclare avoir participé à un comité scientifique pour Cytovation.
Bibliographie
1. Else T, Kim AC, Sabolch A et al. Adrenocortical carcinoma. Endocr Rev 2014 ; 35 : 282‑326.
2. Libé R, Haissaguerre M, Renaudin K et al. Recommandations conjointes du réseau national ENDOCAN-COMETE, de l’Association francophone de chirurgie endocrinienne et de la Société française d’urologie pour la prise en charge du carcinome corticosurrénalien. Bull Cancer 2023 ; 110 : 707‑30.
3. Fassnacht M, Dekkers OM, Else T et al. European Society of Endocrinology Clinical Practice Guidelines on the management of adrenocortical carcinoma in adults, in collaboration with the European Network for the Study of Adrenal Tumors. Eur J Endocrinol 2018 ; 179 : G1‑46.
4. Fassnacht M, Assie G, Baudin E et al. Adrenocortical carcinomas and malignant phaeochromocytomas: ESMO-EURACAN Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2020 ; 31 : 1476‑90.
5. Ghosh C, Hu J, Kebebew E. Advances in translational research of the rare cancer type adrenocortical carcinoma. Nat Rev Cancer 2023 ; 23 : 805‑24.
6. Terzolo M, Fassnacht M, Perotti P et al. Adjuvant mitotane versus surveillance in low-grade, localised adrenocortical carcinoma (ADIUVO): an international, multicentre, open-label, randomised, phase 3 trial and observational study. Lancet Diabetes Endocrinol 2023 ; 11 : 720‑30.
7. Kimpel O, Bedrose S, Megerle F et al. Adjuvant platinum-based chemotherapy in radically resected adrenocortical carcinoma: a cohort study. Br J Cancer 2021 ; 125 : 1233‑8.
8. Fassnacht M, Terzolo M, Allolio B et al. Combination chemotherapy in advanced adrenocortical carcinoma. N Engl J Med 2012 ; 366 : 2189‑97.
9. Campbell MT, Balderrama-Brondani V, Jimenez C et al. Cabozantinib monotherapy for advanced adrenocortical carcinoma: a single-arm, phase 2 trial. Lancet Oncol 2024 ; 25 : 649‑57.
10. Cosentini D, Badalamenti G, Grisanti S et al. Activity and safety of temozolomide in advanced adrenocortical carcinoma patients. Eur J Endocrinol 2019 ; 181 : 681‑9.
11. Sperone P, Ferrero A, Daffara F et al. Gemcitabine plus metronomic 5-fluorouracil or capecitabine as a second-/third-line chemotherapy in advanced adrenocortical carcinoma: a multicenter phase II study. Endocr Relat Cancer 2010 ; 17 : 445‑53.
12. Raj N, Zheng Y, Kelly V et al. PD-1 blockade in advanced adrenocortical carcinoma. J Clin Oncol 2019 ; 38 : 71‑80.
13. Zhu YC, Wei ZG, Wang JJ et al. Camrelizumab plus apatinib for previously treated advanced adrenocortical carcinoma: a single-arm phase 2 trial. Nat Commun 2024 ; 15 : 10371.
14. Baudin E, Grisanti S, Fassnacht M et al. 2MO EO2401 (EO) therapeutic vaccine for patients (pts) with adrenocortical carcinoma (ACC) and malignant pheochromocytoma/paraganglioma (MPP): Phase I/II SPENCER study. Ann Oncol 2022 ; 33 : S545‑6.