Les sarcomes sont des tumeurs rares d’origine mésenchymateuse, représentant moins de 1 % de l’ensemble des tumeurs malignes. Ils peuvent survenir dans n’importe quel site et à tout âge. En outre, il existe une importante hétérogénéité au sein des sarcomes, avec plus de 150 sous-types décrits dans la dernière classification de l’Organisation mondiale de la santé (OMS). En effet, aux divers sous-types histologiques s’ajoute l’identification de sous-types moléculaires distincts, grâce à un accès facilité aux outils de diagnostic moléculaires. Ainsi, même si les sarcomes sont déjà considérés comme rares (www.rarecancereurope.org), ce démembrement histologique et moléculaire crée des maladies encore plus rares.
Malgré les progrès dans la prise en charge locale des sarcomes, notamment via les recommandations de chirurgie en centres experts du réseau NETSARC (https://expertisesarcome.org), les progrès dans les situations avancées/métastatiques sont encore insuffisants, en particulier parce que les précédentes études cliniques ont souvent inclus des patients atteints de sarcomes sans sélections histologiques et/ou moléculaires.
Dans cette revue, nous aborderons les actualités des traitements médicaux des sarcomes, ce qui nous permettra de discuter des progrès récents dans la caractérisation moléculaire et des nouvelles approches thérapeutiques ciblées.
Résumé
Les sarcomes représentent un groupe hétérogène de tumeurs malignes, comprenant plus de 150 sous-types histologiques et moléculaires selon la dernière classification OMS. La plupart de ces tumeurs sont rares (incidence inférieure à 6/100 000/an), voire “ultra-rares” (incidence inférieure à 1/100 000/an). Malgré leur rareté, de nombreux essais cliniques dédiés à certains sous-types histologiques et/ou moléculaires ont été réalisés au cours des dernières années, permettant ainsi l’arrivée de nouveaux médicaments. Dans cette revue, nous nous sommes intéressés aux nouveaux traitements médicaux des sarcomes dont les données ont été publiées et/ou communiquées récemment lors des principaux congrès d’oncologie. Sont ainsi mises en avant des thérapies ciblées (dont des inhibiteurs de tyrosine kinase) ainsi que de l’immunothérapie dans plusieurs sous-types histologiques et moléculaires.
Abstract
News in medical treatments for sarcomas
Sarcomas gather a heterogeneous group of malignant tumours, with more than 150 histological and molecular subtypes according to the latest WHO classification. Most sarcomas subtypes are rare (incidence <6/100.000/year) or even “ultra-rare” (incidence <1/100.000/year). Despite their rarity, a large number of new clinical trials dedicated to histological and/or molecular subtypes have been carried out in the recent years, leading to the use of new drugs. In this review, we focused on new medical treatments for sarcomas with data that have been recently published and/or reported at oncology meetings. We highlight targeted therapies (including tyrosine kinase inhibitors) as well as immunotherapy developed in several histological and molecular subtypes.
Les sarcomes des tissus mous
Avec l’émergence des techniques de biologie moléculaire, nous avons assisté au démembrement moléculaire des sarcomes des tissus mous (STM). Ainsi, plusieurs grands groupes de STM sont identifiés en fonction du type d’anomalie moléculaire qui les caractérise. Outre l’aide au diagnostic, la découverte de ces anomalies moléculaires peut également avoir des conséquences thérapeutiques.
Les liposarcomes
Les liposarcomes bien différenciés et dédifférenciés représentent 15 % de l’ensemble des sarcomes. Ils présentent une amplification constante du gène MDM2, responsable du processus tumoral. En inhibant P53, MDM2 augmente la survie cellulaire.
• Le brigimadlin, inhibiteur de MDM2, a été testé dans une phase Ia/b incluant des liposarcomes dédifférenciés (1) avec une médiane de survie sans progression de 7,9 mois [4,2-9,9] et l’étude de phase III versus doxorubicine en première ligne métastatique vient de clore aux inclusions (NCT05218499).
Le fibrosarcome infantile
Le fibrosarcome infantile, tumeur maligne rare qui survient habituellement au cours de la première année de vie, est caractérisé par une translocation oncogénique ETV6::NTRK3, responsable de la suractivation du récepteur à tyrosine kinase TRK.
• Le larotrectinib est un inhibiteur de tyrosine kinase (ITK) ciblant TRK et dont l’efficacité majeure a été démontrée dans une étude non comparative de phase I/II chez des patients avec cancer avancé et fusion NTRK (2). Sur un total de 159 patients, le taux de réponse objective était de 79 % [72-85], dont 16 % de réponse complète (Fig. 1).
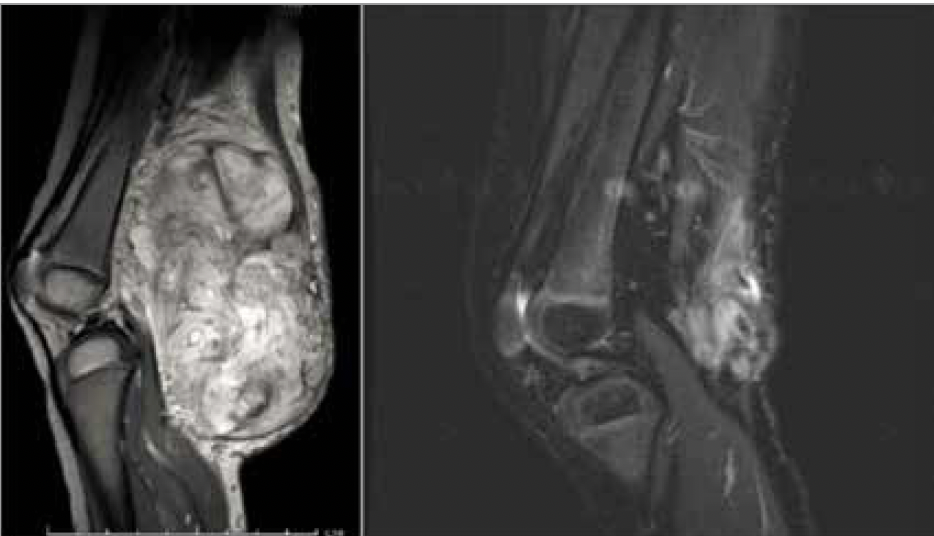
Figure 1 – Fille de 2 ans atteinte d’un fibrosarcome infantile en progression sous chimiothérapie (vincristine actinomycine cyclophosphamide).
En alternative à l’amputation, un essai thérapeutique avec le larotrectinib a été proposé, ayant permis une excellente réponse tumorale : une chirurgie conservatrice avec sur la pièce opératoire une réponse complète pathologique.
Cette étude a abouti en France à une AMM du larotrectinib pour les cas de fibrosarcomes infantiles en situation de rechute et/ou réfractaire à la chimiothérapie ainsi que pour les autres STM pédiatriques avec fusion du gène NTRK, en cas d’échec des traitements standard.
Le sarcome épithélioïde
Le sarcome épithélioïde est un sous-type rare de STM caractérisé par la perte du gène INI1 (également appelé SMARCB1), un gène suppresseur de tumeurs. Cette perte entraîne un déséquilibre du complexe de remodelage de la chromatine SWI/SNF et ainsi une suractivation d’EZH2 à l’origine de la tumorigenèse.
• Le tazémétostat est une thérapie ciblée, modulateur de l’épigénétique, inhibant EZH2. Il a été testé dans une étude de phase II (3) incluant 62 patients avec un sarcome épithélioide métastatique. Le taux de réponse objective était de 15 % [7-26], la médiane de survie sans progression (SSP) de 5,5 mois [3,4-5,9] et la médiane de survie globale de 19 mois [11-non atteinte].
Suite à ces résultats, le tazémétostat est disponible en accès précoce en France dans cette indication.
Les PEComes
Les PEComes sont des sarcomes “ultra-rares” dont le mécanisme d’oncogenèse se fait majoritairement par perte d’hétérozygotie (LOH) du gène TSC2 avec activation de la voie mTOR.
• Le nabsirolimus, thérapie ciblée inhibiteur de mTOR, a été étudié dans une étude de phase II simple bras chez les patients atteints de PEComes avancés/métastatiques (4). Sur 34 patients inclus, le taux de réponse objective était de 39 % [22-58], la médiane de SSP de 10,6 mois [5,5-non atteinte] et la médiane de survie globale (SG) de 40,8 mois [22,2-non atteinte].
Les résultats de cette étude ont permis l’obtention d’une AMM aux États-Unis le 22 novembre 2021.
L’immunothérapie
Par ailleurs, l’immunothérapie a beaucoup été développée dans les STM.
Les inhibiteurs de point de contrôle immunitaire
Dans la majorité des études testant les inhibiteurs de point de contrôle immunitaire (ICI), les patients n’étaient pas sélectionnés et les inclusions ont concerné principalement les sous-types histologiques les plus fréquents (sarcomes indifférenciés pléomorphes, léïomyosarcomes, liposarcomes, etc.). Les résultats des différentes études ont été dans l’ensemble décevants, si bien qu’à l’heure actuelle, aucun ICI ne dispose d’AMM dans cette indication en Europe. Néanmoins, des signaux d’efficacité ont été démontrés dans certains sous-types “ultra-rares”, notamment les sarcomes alvéolaires des parties molles (ASPS).
• Ainsi, une étude de phase II présentée à l’ASCO en 2021 a testé l’atézolizumab chez 44 patients avec un ASPS métastatique (5). Le taux de réponse était de 37,2 % et la médiane de durée de réponse de 16,5 mois [4,9-38,1].
Suite à ces résultats, la FDA a délivré une AMM aux États-Unis pour l’atézolizumab dans les ASPS métastatiques.
• En France, le programme « AcSé Pembrolizumab » d’Unicancer a confirmé l’efficacité du pembrolizumab dans certains sous-types rares de sarcomes, dont les ASPS (6).
• D’autre part, il a été démontré que les structures lymphoïdes tertiaires (TLS) étaient associées à de meilleurs résultats après traitement par ICI dans les sarcomes des tissus mous. Cela a pu être mis en évidence à la fois de manière rétrospective puis prospective dans l’étude PEMBROSARC (7).
Il s’agit donc d’un nouveau biomarqueur potentiel pour sélectionner les patients avec un STM susceptibles de bénéficier des ICI.
La thérapie cellulaire adoptive
La thérapie cellulaire adoptive est une autre forme d’immunothérapie qui consiste en l’utilisation de lymphocytes T cytotoxiques autologues à des fins thérapeutiques (cellules T modifiées avec TCR spécifique ou « TCR-T cells »). Cette technique est prometteuse dans certains sous-types de sarcomes, notamment les synovialosarcomes. Représentant environ 5 à 10 % des STM, ils sont caractérisés par un transcrit de fusion oncogénique SSX::SS18 suspecté d’être à l’origine de l’expression de cancer testis antigen (CTA) comme MAGE-A4. Ces CTA ne sont généralement pas exprimés dans les tissus normaux (en dehors de la période d’embryogenèse).
• Les résultats d’une étude de phase II testant l’utilisation de l’afami-cel (TCR T cells anti-MAGE-A4) chez des patients avec un synovialosarcome métastatique exprimant MAGE-A4 ont été présentés à l’ASCO. Le taux de réponse était de 39,4 % et le taux de contrôle de la maladie de 84,8 % (8).
Les sarcomes osseux
Le sarcome d’Ewing
• Dans le sarcome d’Ewing, l’étude Euro Ewing 2012 a été conçue notamment pour comparer le protocole européen (induction par VIDE et consolidation par VAI ou VAC ou chimiothérapie haute dose busulfan/melphalan en fonction du risque de progression des patients) et du protocole américain (induction dose-dense par VDC/IE et consolidation par IE/VC) et ce, en termes d’efficacité et de tolérance. Elle a inclus 640 patients avec un sarcome d’Ewing nouvellement diagnostiqué localisé ou métastatique d’âge entre 5 et 50 ans. Avec un suivi médian de 1,7 an, la SSP et la SG étaient en faveur du protocole américain (HR = 0,7 ; IC 95 % = 0,51-0,95 et HR = 0,64 ; IC 95 % = 0,42-0,96). Cette supériorité est retrouvée dans tous les sous-groupes de patients (sexe, âge, stade de la maladie, volume tumoral et pays). Les toxicités étaient comparables dans les deux groupes thérapeutiques.
Le schéma VDC/IE est donc le nouveau standard international de la prise en charge initiale des sarcomes d’Ewing.
Les ostéosarcomes
En situation de maladie récidivante et/ou réfractaire à la chimiothérapie, le pronostic des ostéosarcomes est sombre, avec des médianes de survie ne dépassant pas les 12 mois. À l’heure actuelle, les médicaments les plus prometteurs sont les ITK multicibles oraux avec activité antiangiogénique.
• L’étude de phase II randomisée REGOBONE (9) a testé le régorafénib versus placebo (avec possibilité de cross-over à la progression) dans cette indication. Avec 65,4 % des patients non progressifs à 8 semaines dans le bras régorafénib contre 0 dans le bras placebo, la médiane de SSP était significativement augmentée dans le bras régorafénib : 13,7 [8,0-27,3] versus 4 semaines [3,0-5,7]. En outre, le taux de survie globale à 1 an était de 53 [31-71] versus 33 % [10-59] en faveur du régorafénib.
• Ce traitement est actuellement évalué en maintenance après une prise en charge thérapeutique standard d’un ostéosarcome de haut grade à la phase initiale de la maladie (NCT04055220) et en première rechute (NCT04698785).
Les GIST
Les tumeurs stromales gastro-intestinales (GIST) sont des tumeurs mésenchymateuses malignes qui peuvent se développer tout le long du tractus digestif. Elles sont caractérisées par des mutations activatrices des gènes KIT ou PDGFRA dans plus de 90 % des cas. Ainsi, plusieurs inhibiteurs de tyrosine kinase (ITK) ciblant KIT et PDGFRA ont été développés dans cette pathologie, dont l’imatinib, le sunitinib et le régorafénib, qui constituent respectivement les traitements de première, deuxième et troisième lignes en situation de maladie avancée/métastatique.
Le riprétinib
Au-delà de la troisième ligne, un nouvel ITK a été développé récemment, le riprétinib. Il s’agit d’un médicament agissant à la fois en bloquant la poche ATP de la kinase, mais également la boucle d’activation de la kinase.
• Le riprétinib a été évalué en quatrième ligne dans une étude de phase III randomisée versus placebo, avec possibilité de cross-over à la progression (10). Cette étude a démontré un bénéfice à la fois en termes de survie sans progression, qui était le critère de jugement principal (1 versus 6 mois ;
p < 0,0001), et en termes de survie globale (6 versus non atteinte), et ce, malgré le cross-over. Dans l’ensemble, le traitement était bien toléré avec principalement répertoriés des effets secondaires de grade I-II (alopécie, nausées, fatigue) (Fig. 2).
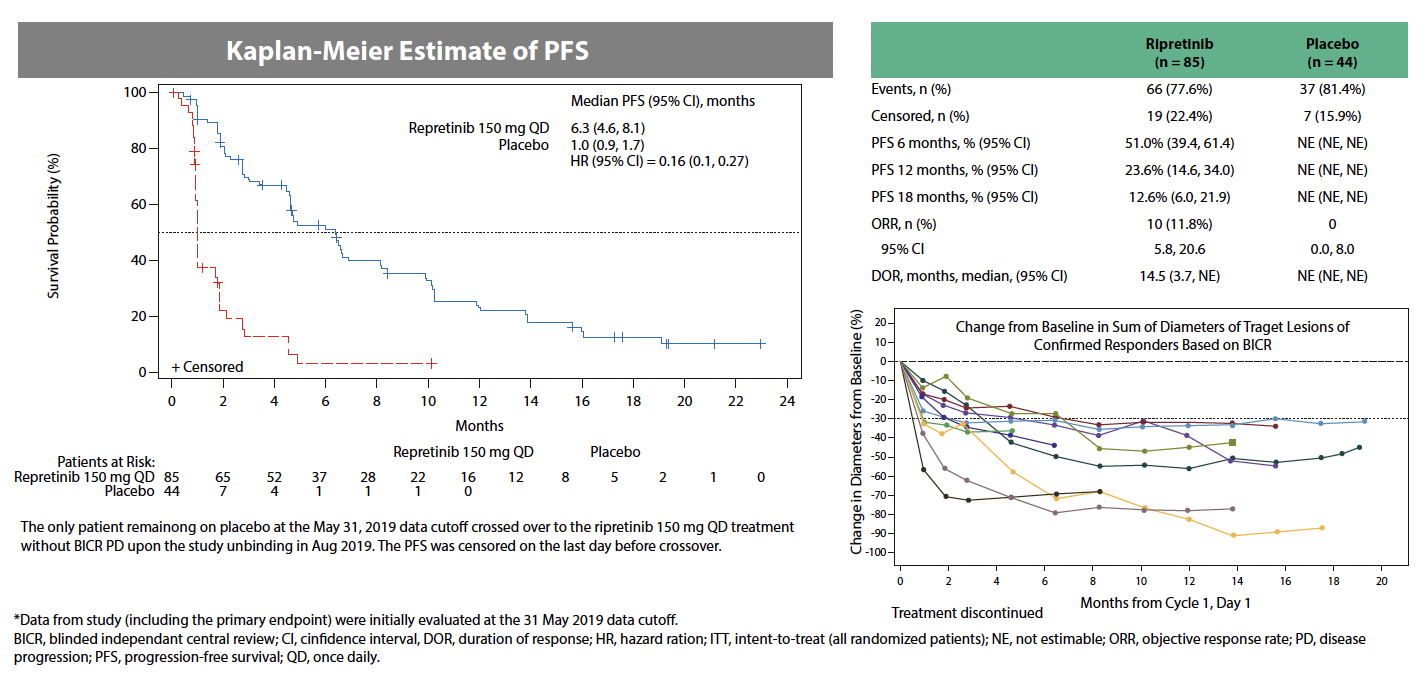
Figure 2 – Courbe de survie sans progression des patients de l’étude INVICTUS-DCC (10).
Les résultats montrent une amélioration significative dans bras riprétinib versus placebo (1 versus 6 mois, p < 0,0001).
Le riprétinib est donc disponible en quatrième ligne dans le cadre d’un accès précoce depuis le
18 février 2022.
L’avapritinib
Par ailleurs, dans moins de 5 % des cas, les GIST sont caractérisées par la mutation D842V de l’exon 18 de PDGFRA, qui est systématiquement résistante à l’imatinib et aux autres ITK ayant l’AMM.
• Ainsi, un ITK ciblant spécifiquement cette mutation a été développé, l’avapritinib. Ce médicament a été testé dans le cadre d’une étude de phase Ia/b (11) avec une efficacité majeure (taux de réponse objective de 91 % (51/56 patients), médiane de survie sans progression de 34 mois) (Fig. 3).
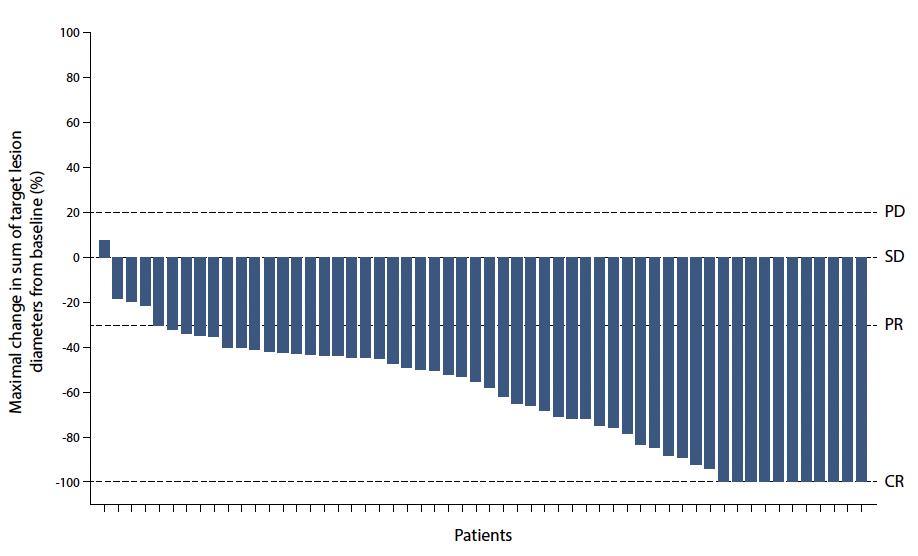
Figure 3 – Diagramme en cascade des patients avec une GIST au stade avancé/métastatique et mutation D842V de l’exon 18 de PDGFRA traités par avapritinib (11).
Le taux de réponse objective est de 91 %.
L’avapritinib a obtenu une AMM depuis le 11 mars 2021. À noter dans le profil de tolérance une toxicité neurocognitive restant mal comprise et pour laquelle les autorités de santé ont donc exigé la mise en place d’un registre pour le suivi de tolérance au long cours.
Les tumeurs conjonctives à malignité intermédiaire
Les tumeurs desmoïdes
Les tumeurs desmoïdes sont des tumeurs conjonctives à malignité intermédiaire, avec risque évolutif locorégional, mais sans potentiel métastatique. Elles peuvent néanmoins devenir invalidantes et avoir une incidence sur la qualité de vie des patients, voire parfois menacer le pronostic vital par envahissement locorégional d’organes vitaux.
• Le nirogacestat est un inhibiteur de la gamma-sécrétase, entraînant une inhibition de la signalisation Notch. Ce traitement a été testé dans une étude de phase III randomisée versus placebo, ayant inclus 142 patients avec une tumeur desmoïde progressive (12). Le critère de jugement principal était la SSP, avec un bénéfice significatif du nirogacestat (SSP non atteinte dans le bras nirogacestat versus 15 mois dans le bras placebo ; HR = 0,29 ; p < 0,001). Le taux de réponse objective était également significativement amélioré par le nirogacestat (41 versus 8 % ; p < 0,001). La tolérance était principalement marquée par des toxicités digestives (diarrhées et/ou nausées) de grade 1 ou 2, avec tout de même 16 % de diarrhées grade 3. À noter également la survenue d’une insuffisance ovarienne chez 75 % des patientes, qui semble résolutive à l’arrêt du traitement, mais dont le mécanisme reste encore mal compris.
À l’heure actuelle, le nirogacestat dispose d’un accès précoce en France.
Les tumeurs à cellules géantes ténosynoviales
Les tumeurs à cellules géantes ténosynoviales (TCGT, anciennement appelées synovites villonodulaires pigmentées) sont également des tumeurs conjonctives à malignité intermédiaire. Dans leur forme diffuse, l’exérèse chirurgicale est suivie de récidive dans près de la moitié des cas et ces tumeurs peuvent être responsables de douleurs et d’une impotence fonctionnelle sévère. D’un point de vue moléculaire, elles sont caractérisées par un transcrit de fusion impliquant le gène CSF1 et à l’origine de la tumorigenèse.
• Le vimseltinib est un ITK ciblant CSF1R. Ce traitement très prometteur a été testé dans étude de phase III randomisée versus placebo (NCT05059262), ayant inclus plus de 120 patients avec une TCGT diffuse (randomisation 2 :1). Les résultats sont attendus pour 2024.
Conclusion
Le développement des outils d’analyse moléculaire a permis d’une part une meilleure compréhension des mécanismes d’oncogenèse et d’autre part une meilleure classification des sarcomes, aboutissant à ce que l’on appelle de “démembrement moléculaire des sarcomes”.
S’en sont suivi de plus en plus d’essais thérapeutiques ciblant un sous-type histologique et moléculaire, avec des résultats plus intéressants que les précédentes études qui incluaient tous les sous-types de sarcomes sans sélection. C’est donc tout naturellement que le paysage thérapeutique des sarcomes s’est étoffé au cours des dernières années, avec plusieurs AMM et AP octroyés récemment à des ITK et à d’autres thérapies ciblées comme les modulateurs épigénétiques. S’agissant de l’immunothérapie, les ICI font enfin leur entrée dans le domaine du sarcome, avec notamment une efficacité remarquable dans certains sous-types très rares comme les ASPS et l’obtention d’une AMM aux États-Unis dans cette indication. De plus, des biomarqueurs prédictifs d’efficacité, toujours à l’étude, semblent pertinents pour mieux sélectionner les sous-types de sarcomes sensibles à l’immunothérapie. Enfin, les nouvelles formes d’immunothérapie, notamment les TCR-T cells, présentent des premiers résultats très encourageants même s’il s’agit à l’heure actuelle de traitements expérimentaux.
Mehdi Brahmi déclare avoir des liens d’intérêt avec Deciphera, Pharmamar et Mundipharma. Axel de Bernardi déclare ne pas avoir de liens d’intérêt. Armelle Dufresne déclare avoir des liens d’intérêt avec GSK, Pharmamar et Mundipharma. Jean-Yves Blay déclare avoir des liens d’intérêt avec Novartis, GSK, Bayer, Roche, Deciphera, Ignyta, BMS, MSD, Pharmamar, Karyopharm et Boehringer Ingelheim.
Bibliographie
1. LoRusso P, Gounder MM, Yamamoto N et al. A phase Ia/Ib, dose-escalation/expansion study of the MDM2–p53 antagonist BI 907828 in patients (pts) with solid tumors: Safety and efficacy in patients with dedifferentiated liposarcoma (DDLPS). J Clin Oncol 2023 ; 41 : 11554.
2. Hong DS, DuBois SG, Kummar S et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol 2020 ; 21 : 531‑40.
3. Gounder M, Schöffski P, Jones RL et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet Oncol 2020 ; 21 : 1423‑32.
4. Wagner AJ, Ravi V, Riedel RF et al. Nab-sirolimus for patients with malignant perivascular epithelioid cell tumors. J Clin Oncol 2021 ; 39 : 3660‑70.
5. Naqash AR, O’Sullivan Coyne GH, Moore N et al. Phase II study of atezolizumab in advanced alveolar soft part sarcoma (ASPS). J Clin Oncol 2021 ; 39 : 11519.
6. Blay JY, Chevret S, Le Cesne A et al. Pembrolizumab in patients with rare and ultra-rare sarcomas (AcSé Pembrolizumab): analysis of a subgroup from a non-randomised, open-label, phase 2, basket trial. Lancet Oncol 2023 ; 24 : 892‑902.
7. Italiano A, Bessede A, Pulido M et al. Pembrolizumab in soft-tissue sarcomas with tertiary lymphoid structures: a phase 2 PEMBROSARC trial cohort. Nat Med 2022 ; 28 : 1199‑206.
8. D’Angelo SP, Van Tine BA, Attia S et al. SPEARHEAD-1: A phase 2 trial of afamitresgene autoleucel (Formerly ADP-A2M4) in patients with advanced synovial sarcoma or myxoid/round cell liposarcoma. J Clin Oncol 2021 ; 39 : 11504.
9. Duffaud F, Mir O, Boudou-Rouquette P et al. Efficacy and safety of regorafenib in adult patients with metastatic osteosarcoma: a non-comparative, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Oncol 2019 ; 20 : 120‑33.
10. Blay JY, Serrano C, Heinrich MC et al. Ripretinib in patients with advanced gastrointestinal stromal tumours (INVICTUS): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 2020 ; 21 : 923‑34.
11. Jones RL, Serrano C, Mehren M von et al. Avapritinib in unresectable or metastatic PDGFRA D842V-mutant gastrointestinal stromal tumours: Long-term efficacy and safety data from the NAVIGATOR phase I trial. Eur J Cancer 2021 ; 145 : 132‑42.
12. Gounder M, Ratan R, Alcindor T et al. Nirogacestat, a γ-secretase inhibitor for desmoid tumors. N Engl J Med 2023 ; 388 : 898‑912.