Résumé
Au cours des 2 dernières décennies, les efforts de recherche ont apporté une mine de connaissances, conduisant à une meilleure compréhension des bases moléculaires des leucémies aiguës pédiatriques. Ceci a permis une stratification de l’intensité du traitement selon les caractéristiques biologiques des cellules leucémiques d’une part et la réponse précoce au traitement d’autre part, tous deux fortement prédictifs du risque de rechute. Ces stratégies de médecine de précision ont conduit à l’amélioration progressive des protocoles de chimiothérapie et au recours à la greffe de cellules souches hématopoïétiques, mais aussi au développement de thérapies innovantes, qu’elles soient dites ciblées ou immunothérapies. Si la survie s’est améliorée, elle s’est aussi accompagnée d’une meilleure prise en compte des effets secondaires précoces et tardifs qui contribue à une meilleure qualité de vie des patients guéris.
Abstract
Acute pediatric leukemias: therapeutic progress over the last 20 years
Over the past two decades, research efforts have provided a huge improvement of knowledge, leading to a better understanding of the molecular basis of acute pediatric leukemia that allowed a stratification of the intensity of treatment according to the biological characteristics of leukemia cells and the early response to treatment, highly predictive of the risk of relapse. These precision medicine strategies have led to the progressive improvement of chemotherapy regimens and of the use of hematopoietic stem cell transplantation but also to the development of innovative therapies, whether they are named targeted or immunotherapies. If survival has improved, a better consideration of early and late side effects has also accompanied the treatments protocols and contribute to a better quality of life for patients cured.
Introduction
Épidémiologie
Les leucémies aiguës représentent environ 30 % de l’ensemble des cancers pédiatriques et sont ainsi la première cause de cancer chez l’enfant de moins de 15 ans. Les leucémies aiguës pédiatriques se répartissent en :
• leucémies aiguës lymphoblastiques (LAL), qui sont majoritaires (près de 80 %)
• et leucémies aiguës myéloïdes (LAM).
On dénombre chaque année environ 400 nouveaux cas de LAL chez les moins de 15 ans en France, se répartissant pour environ 85 % d’entre elles en LAL de la lignée B (LAL-B) et 15 % en LAL de la lignée T (LAL-T). Le pic d’incidence se situe entre l’âge de 2 et 5 ans (1). Parallèlement, 65 à 85 enfants de moins de 15 ans vont présenter chaque année une LAM, sans pic d’âge de survenue, hormis les 2 premières années de vie où l’incidence est supérieure.
Pronostic
La survie des enfants atteints de leucémie (Fig. 1 et 2) s’est améliorée régulièrement depuis 50 ans, essentiellement grâce aux connaissances issues de la recherche clinique, mais l’enjeu des prochaines décennies sera de guérir les formes en rechute ou réfractaires, tout en s’intéressant aux effets secondaires précoces et tardifs afin d’améliorer au mieux la qualité de vie des patients guéris (2, 3).
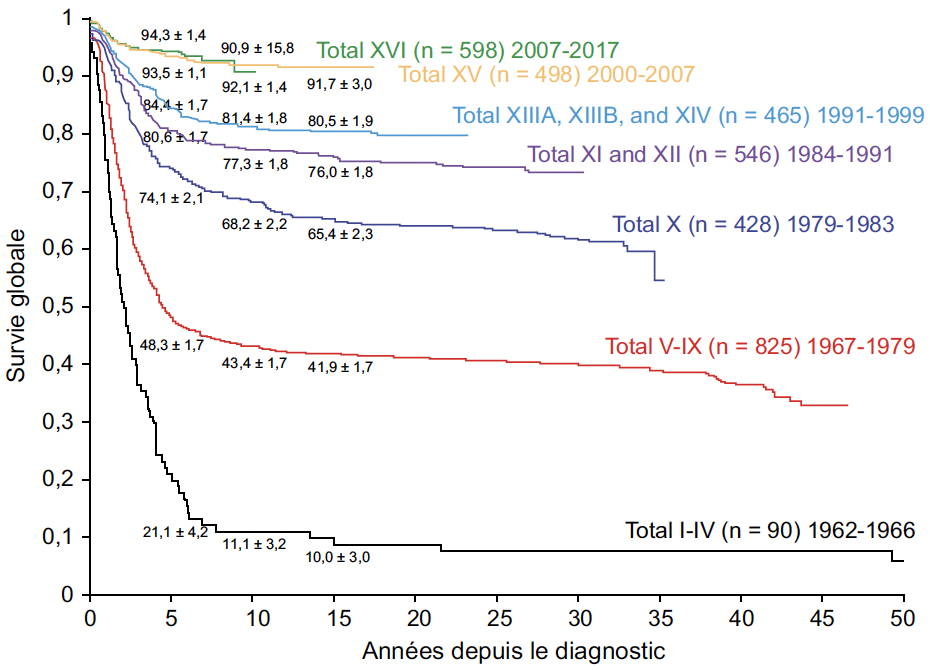
Figure 1 – Progression de la survie globale des patients traités pour une LAL pédiatrique ces 40 dernières années par les protocoles du St-Jude (d’après 2).
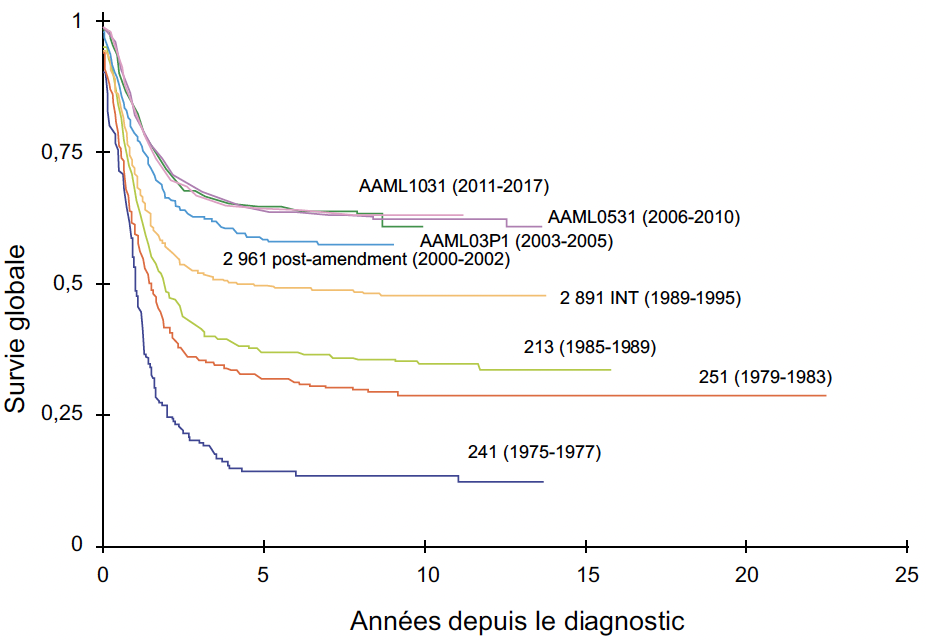
Figure 2 – Progression de la survie globale des patients traités pour une LAM pédiatrique ces 40 dernières années par les protocoles du COG (d’après 3).
Facteurs de risque
Nous n’aborderons pas dans cet article les facteurs de risque de survenue d’une leucémie aiguë dans l’enfance, cependant nous savons désormais que le processus de leucémogenèse n’est pas dépendant d’un seul événement causal, mais est le résultat de l’interaction entre un terrain génétique constitutionnel particulier et divers événements environnementaux et génétiques secondaires pouvant survenir au cours du développement de l’hématopoïèse ante et postnatale.
De nouvelles données moléculaires
Les leucémies aiguës lymphoblastiques
Les LAL sont des pathologies clonales hétérogènes caractérisées par des altérations génétiques somatiques pouvant survenir à différents stades du développement lymphoïde. L’identification de ces événements génétiques récurrents, mutuellement exclusifs, est d’un intérêt capital pour la prise en charge des patients puisqu’elle a un effet direct sur le pronostic et la stratification thérapeutique.
Les LAL-B
Grâce au développement récent des techniques de profilage moléculaire (new generation sequencing – NGS) (4), un nouveau paysage génomique des LAL-B a été mis en lumière et l’on est aujourd’hui capable de retrouver une anomalie génétique “classante” chez plus de 90 % des enfants atteints.
• Ces anomalies initiatrices concernent des variations de la ploïdie (gain ou perte de chromosomes entiers), des translocations chromosomiques qui dérégulent les gènes et impliquent généralement des facteurs de transcription hématopoïétiques ou des facteurs épigénétiques ou encore des récepteurs de cytokines ou de kinases.
• À ces événements initiateurs peuvent s’associer des variations du nombre de copies des gènes (gain ou perte) et des mutations ponctuelles dans de multiples voies cellulaires. De nouvelles entités ont ainsi été décrites avec la mise en évidence de réarrangements qui impliquent un même gène (MEF2D et ZNF384 par exemple), mais avec de multiples partenaires ou sont cryptiques par analyse cytogénétique classique (par exemple avec DUX4).
• D’autres sous-types de LAL peuvent être caractérisés par divers gènes initiateurs (par exemple LAL Ph-like) ou sont caractérisés par des mutations de séquence (par exemple PAX5 P80R et IKZF1 N159Y).
• D’autres groupes, enfin, ont des profils d’expression génétique similaires à ceux de sous-types connus avec différentes altérations génétiques (Ph-like et ETV6::RUNX1-like ALL) (Fig. 3A). Ces anomalies ont de surcroît une fréquence différente chez l’enfant et l’adulte et des pronostics différents (Fig. 3B).
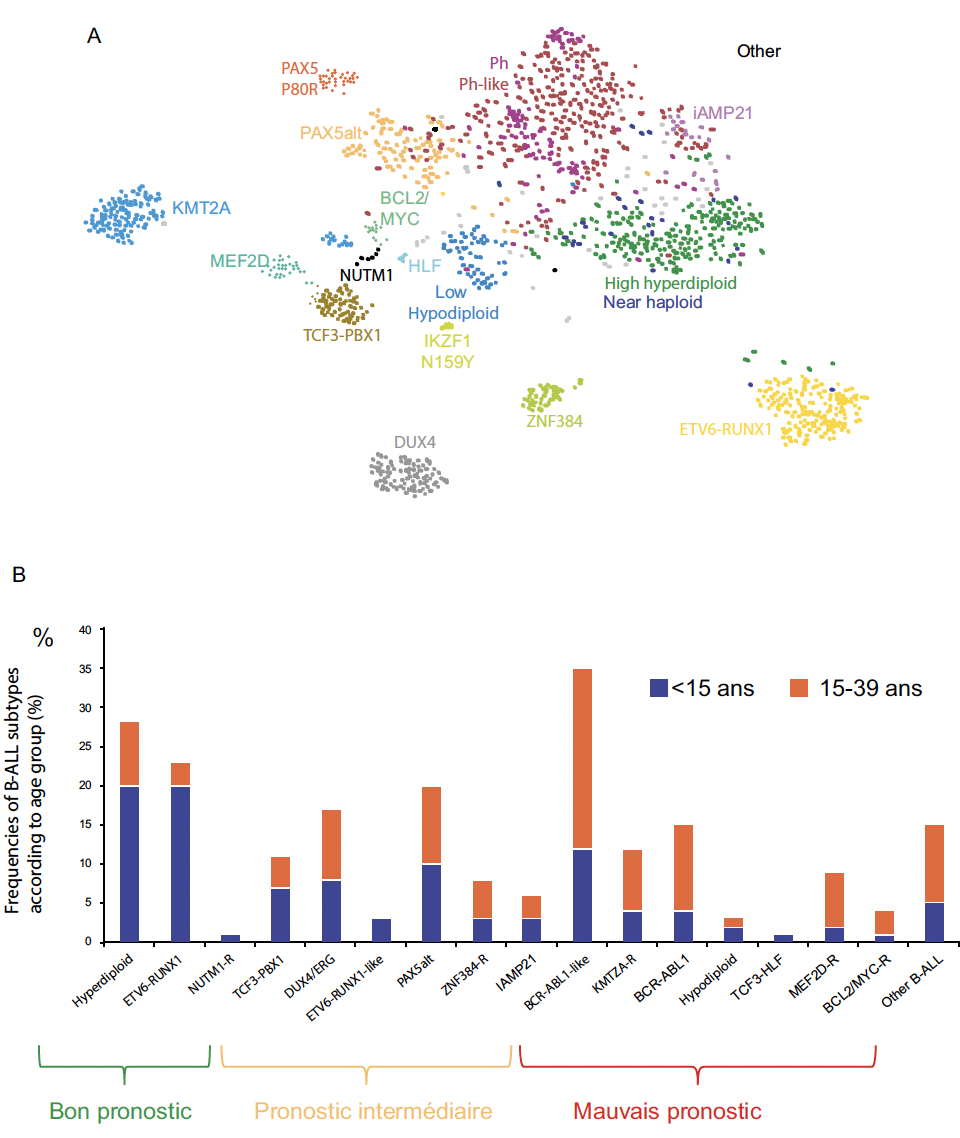
Figure 3 – A : Représentation par tSNE (t-distributed stochastic neighbor embedding) des sous-types de LAL-B fondée sur les profils d’expression en RNASeq de 1 988 cas. Cette méthode de réduction de dimension permet de regrouper des données proches (d’après 5).
B : Fréquence des sous-types génétiques de LAL-B selon le groupe d’âge (d’après 6).
Les LAL-T
Contrairement aux LAL-B, les altérations génétiques sentinelles des LAL-T n’ajoutent pas de valeur pronostique significative pour affiner la stratification des risques, mais plusieurs anomalies mettent en évidence des opportunités potentielles de thérapie ciblée.
• La voie Notch est la voie de signalisation la plus couramment dérégulée dans les LAL-T, chez environ 65 % des patients, soit par une mutation activatrice de NOTCH1 (~ 50 %), soit par une perte de fonction du régulateur négatif FBXW7 (~ 15 %).
• La voie de signalisation PI3K/AKT/m-TOR est activée dans environ 30 % des cas LAL-T, principalement à partir de mutations ou de délétions de PTEN, conduisant à l’activation d’AKT. Cette voie peut également être activée directement par des mutations dans PIK3CA, PIK3R1, IL7R ou AKT ou indirectement par des mutations de la voie Notch, Ras et JAK-STAT.
• Des mutations de la voie JAK-STAT et Ras se produisent respectivement dans environ 25 % et 15 % des cas de LAL-T et sont particulièrement présentes dans les formes à précurseurs T précoces (ETP) (7).
Les leucémies aiguës myéloïdes
Les LAM pédiatriques ont aussi bénéficié des approches NGS permettant d’identifier des translocations cryptiques ou non détectées auparavant (Tab. 1). Il s’agit, par exemple de CBFA2T3::GLIS2 ou des fusions de NUP98.
Stratification des risques et marqueurs pronostiques, dont la maladie résiduelle
L’intégration des données moléculaires
L’intégration de ces données moléculaires dans les algorithmes de stratification des risques est devenue essentielle dans les LAL comme dans les LAM et s’est ajoutée aux critères péjoratifs clinico-biologiques précédemment utilisés pour les LAL :
• âge (< 1 an ou > 10 ans),
• leucocytose sanguine au diagnostic (> 50 x 109/l),
• atteinte neuroméningée,
• immunophénotype T.
Dans les LAL-B
L’un des premiers exemples repose sur l’identification des altérations d’IKZF1 en tant que marqueurs pronostiques défavorables dans les LAL-B qui a conduit à des approches de traitement adaptées (9). La plupart des altérations d’IKZF1 sont retrouvées dans les LAL BCR::ABL1 et les Ph-like, mais leur présence dans les LAL DUX4 réarrangées n’est pas de mauvais pronostic. Cela a mené à la définition récente d’une catégorie « IKZF1-plus » de mauvais pronostic, qui est définie par la présence d’altérations d’IKZF1 et de CDKN2A/B, PAX5 ou PAR1 (comme marqueur des réarrangements CRLF2 non repérés par les techniques de MLPA), sans altération d’ERG (marqueur des réarrangements de DUX4 non repérés en MLPA).
Dans les LAM
Pour les LAM, les analyses génétiques et moléculaires ciblées définissent aussi un risque “génétique” qui incluent des anomalies connues de longue date comme la monosomie 5/del(5q), la monosomie 7 ou les anomalies en 12p et des plus récentes, comme les translocations CBFA2T3::GLIS2, DEK::NUP214, les fusions NUP98, et les réarrangements de MLLT10 ou les mutations de RUNX1, WT1 et PHF6 qui représentent des pronostics défavorables avec les chimiothérapies conventionnelles.
La réponse précoce à la chimiothérapie définie par la maladie résiduelle
La réponse précoce à la chimiothérapie définie par la maladie résiduelle (MRD : Minimal Residual Disease) est le deuxième facteur pronostique fondamental (10).
Elle peut être mesurée par cytométrie en flux (dans les LAL comme dans les LAM), identifiant un profil phénotypique aberrant spécifique à la leucémie appelé LAIP (Leukemia Associated ImmunoPhenotype) ou par PCR quantitative des gènes des chaînes lourdes des immunoglobulines ou du TCR spécifiques à la leucémie (LAL) ou encore des transcrits de fusion. Les nouvelles techniques de détection (NGS ou digital PCR) sont plus sensibles, mais leur apport en clinique reste à confirmer. Plus de 95 % des leucémies aiguës pédiatriques pourront ainsi bénéficier d’un suivi de la MRD.
En pratique
En pratique, l’évaluation du risque de rechute, et donc l’intensité du traitement finalement proposé, reposera sur la combinaison des caractéristiques génétiques/moléculaires de la leucémie et de la réponse précoce du patient (en fin d’induction et/ou de consolidation) au traitement entrepris.
Globalement pour les LAL, le risque d’échec du traitement est de trois à cinq fois plus élevé chez les enfants avec des niveaux de MRD ≥ 10-4 (0,01 %) en fin d’induction. Les protocoles les plus récents intègrent même dans leur algorithme de stratification des seuils pronostiques différents selon le sous-type génétique de la LAL-B (ex. : protocole ALLTogether EUDRACT n° 2018-001795-38).
La greffe de cellules souches hématopoïétiques
L’allogreffe de cellules souches hématopoïétiques
L’allogreffe de cellules souches hématopoïétiques (CSH) offre un traitement potentiellement curatif grâce à l’effet combiné d’un conditionnement préparatoire intense et de l’effet du greffon contre leucémie (GvL). Au fil des ans, les progrès en matière de typage HLA en haute résolution des donneurs, le choix du conditionnement, la prophylaxie de la maladie du greffon contre l’hôte (GvH) et les mesures de soins de support ont continuellement amélioré les résultats de la greffe en termes de survie.
Pour autant, compte tenu de l’efficacité grandissante des traitements médicamenteux (chimiothérapie, immunothérapie et thérapies ciblées) et du risque potentiel de toxicité aiguë et tardive après la greffe de CSH, la pratique actuelle limite l’utilisation de celle-ci, en pédiatrie, aux formes à risque élevé de rechute que ce soit dans les LAL ou les LAM.
Les conditionnements
Dans cet esprit, les conditionnements à toxicité atténuée ne sont pas utilisés, sauf en cas de comorbidité élevée. Si, dans les LAM, l’utilisation des conditionnements avec irradiation corporelle totale (ICT) n’a pas montré de bénéfice par rapport à des conditionnements ne comportant que de la chimiothérapie, l’essai randomisé FORUM européen a démontré la supériorité en termes de survie post-greffe des conditionnements avec ICT dans les LAL, pourtant plus compliqués à mettre en œuvre et plus pourvoyeurs de séquelles à long terme. En effet, cet essai a prouvé un gain de survie de 20 % pour l’ICT comparativement aux conditionnements myéloablatifs fondés sur du busulfan ou du tréosulfan et dans tous les sous-groupes (11).
Les greffons alternatifs
Parallèlement le développement de greffons alternatifs rend désormais cette thérapie cellulaire quasiment accessible à tous les patients qui en relèvent. En effet, les greffes de sang placentaire qui ont vu leur essor dans le début des années 2000 sont désormais de plus en plus remplacées par les greffes haplo-identiques intrafamiliales utilisant le cyclophosphamide post-greffe. Cette prophylaxie puissante de la GvH est en effet facilement transposable partout, contrairement aux techniques de sélection cellulaire développées initialement selon le protocole de F. Aversa (12).
La maladie résiduelle avant et après greffe
En outre, la maladie résiduelle avant et après greffe est de plus en plus finement prise en compte et des interventions afin de favoriser l’effet GvL peuvent être utilisées pour améliorer la survie sans leucémie après allogreffe. Cet objectif a été poursuivi au moyen de plusieurs stratégies, notamment l’arrêt rapide de l’immunosuppression post-greffe et la perfusion de lymphocytes dérivés du donneur ou d’effecteurs immunitaires stimulés par cytokine. Le concept de traitement d’entretien post-greffe a aussi vu le jour avec le développement des thérapies ciblées comme les inhibiteurs de la tyrosine kinase qui sont recommandés dans les 1 à 2 ans post-greffe pour les LAL Philadelphie (protocole EsPhALL 2017/COG AALL1631 n° 2017-000705-20).
Les thérapies ciblées
Les schémas de chimiothérapie adaptés aux facteurs de risque génétiques et à la maladie résiduelle n’ont pas permis d’améliorer le pronostic de tous les enfants, en particulier les adolescents et les nourrissons ou les enfants en rechute/réfractaires, et ceci, malgré des essais cliniques randomisés menés par des consortiums de collaboration internationale. Néanmoins, les découvertes génomiques réalisées au cours des 2 dernières décennies offrent le rationnel biologique à une médecine de précision fondée sur les vulnérabilités mises en évidence par les techniques de NGS.
Dans les LAL Ph
La première thérapie fondée sur les caractéristiques génomiques des LAL-B a été utilisée pour le traitement des LAL à chromosome Philadelphie (Ph) avec transcrit BCR::ABL1. Les LAL Ph étaient historiquement associées à un pronostic défavorable et l’allogreffe de CSH était la meilleure option curative avant l’ère des inhibiteurs de tyrosine kinase (ITK).
• Depuis les années 2010, l’adjonction précoce d’ITK (imatinib puis ITK de 2e voire 3e génération) a permis non seulement un gain de survie de 20 %, mais aussi de réduire le recours à l’allogreffe, qui concerne désormais moins de 20 % des enfants (6).
Dans les LAL Ph-like
La découverte récente d’un groupe de LAL dites Ph-like a étendu le champ des thérapies ciblées, ce, d’autant que si les LAL Ph concernent 3 à 5 % des enfants, les LAL Ph-like surviennent chez 15 % d’entre eux, et particulièrement chez les adolescents (Fig. 3). Ce groupe hétérogène au plan des altérations génomiques conduit à un profil d’expression génique proche des LAL Ph d’où son intitulé. Les diverses anomalies qui le composent convergent vers l’activation des voies de signalisation JAK-STAT, ABL ou Ras/MAPK, chacune d’entre elles pouvant être inhibée par un ITK adéquat.
• Plusieurs essais sont en cours pour évaluer l’efficacité de ces ITK en association avec la chimiothérapie dans les LAL présentant des fusions de la classe ABL (avec l’imatinib, dasatinib ou ponatinib) ou de NTRK (avec le larotrectinib) et des altérations de la voie JAK-STAT (avec le ruxolitinib) (6).
Dans les LAL du nourrisson
Les LAL du nourrisson sont aussi un enjeu pour ces nouvelles thérapies.
• Plusieurs pistes sont explorées avec les inhibiteurs de l’interaction menin-KMT2A, des inhibiteurs de DOT1L, qui est une histone méthyltransférase liée à plusieurs partenaires de KMT2A, ou encore des modulateurs épigénétiques comme les agents déméthylants (azacitidine).
• Le vénétoclax, un inhibiteur sélectif de BCL2, est aussi en expérimentation clinique dans cette indication.
Dans les LAL-T
Les LAL-T ont pour l’instant moins bénéficié de l’exploitation de leurs vulnérabilités génomiques.
• En effet, les premières recherches ont porté sur la voie NOTCH, dérégulée dans 65 % des LAL-T, mais les inhibiteurs de première génération avaient malheureusement une toxicité digestive limitante.
• Les efforts se sont alors portés sur les inhibiteurs de la voie PI3K/AKT/mTOR avec l’évérolimus par exemple ou, pour les LAL avec mutations de la voie Ras, le sélumétinib, mais restent préliminaires.
• D’autres opportunités de ciblage sont en cours sur les gènes du cycle cellulaire ou ceux des régulateurs épigénétiques.
Dans les LAM
• Enfin dans les LAM, les inhibiteurs de l’apoptose BCL2 médiée comme le vénétoclax ont montré leur efficacité dans les rechutes et LAM réfractaires en combinaison avec des agents hypométhylants et de la chimiothérapie à faible dose. Leur place en première ligne reste à déterminer.
• Les autres thérapies ciblent essentiellement FLT3, avec actuellement des inhibiteurs plus sélectifs comme le gilteritinib ou le quizartinib ou les inhibiteurs de menin (révuménib).
• Les inhibiteurs du protéasome, comme le bortézomib, n’ont pas fait preuve de leur efficacité dans les essais cliniques, mais l’effet du pévonédistat, un inhibiteur de NEDD8, en association avec la chimiothérapie, est en cours d’analyse.
• Enfin, des molécules ciblant la régulation épigénétique sont en cours d’investigation comme les inhibiteurs de méthyltransférase (azacitidine ou décitabine), les inhibiteurs d’histone désacétylase (vorinostat, panobinostat) ou encore des inhibiteurs d’IDH1/2 (ivosidénib, énasidénib).
L’immunothérapie
L’immunothérapie peut être administrée sous forme d’anticorps plus ou moins humanisés ou de thérapie cellulaire T (cellules T modifiées pour exprimer un récepteur à l’antigène chimérique, CAR-T cells). Les anticorps monoclonaux ciblent un cluster de différentiation (CD) à la surface de la cellule leucémique et doivent ensuite être capables de provoquer un effet cytotoxique direct ou grâce à une molécule toxique couplée à l’anticorps.
Les anticorps monoclonaux
Le rituximab
Le rituximab, anticorps anti-CD20, a été le premier utilisé dans les années 1990, mais son adjonction à la chimiothérapie dans les LAL-B de l’enfant n’a pas permis d’améliorer les courbes de survie déjà très hautes.
L’inotuzumab ozogamicine
L’inotuzumab ozogamicine est un anticorps anti-CD22 (présent sur 90 % des LAL-B) couplé à la calichéamicine, puissant agent cytotoxique. Son utilisation s’est développée en alternative ou en association à la chimiothérapie dans les LAL-B en rechute et fera partie du prochain protocole thérapeutique de phase III européen du consortium IntREALL. Sa demi-
vie longue lui confère un atout thérapeutique, mais sa toxicité hépatique (syndrome d’occlusion sinusoïdale) incite à le positionner très en amont de la réalisation d’une allogreffe de CSH.
L’épratuzumab
Un autre anticorps anti-CD22, l’épratuzumab, a été testé dans les années 2010 en complément de la chimiothérapie, mais il n’a pas fait preuve de son efficacité et son développement s’est arrêté (2).
Le blinatumomab
Ces dernières années ont vu en revanche l’avènement d’un anticorps bispécifique anti-CD19/CD3. Le blinatumomab est composé de deux fragments variables différents, l’un reconnaissant le CD3 et permettant d’activer la cytotoxicité T et, l’autre, le CD19, qui est présent dans la quasi-totalité des LAL-B. Cet anticorps nécessite une administration intraveineuse continue et l’association à une prophylaxie neuroméningée classique, car sa pénétration cérébrale est faible bien qu’il soit pourvoyeur d’une toxicité neurologique. Au fil du temps son positionnement dans l’arsenal thérapeutique s’est affiné depuis que l’essai IntREALL HR 2010 (NCT03590171), dans lequel il était introduit en consolidation, a montré son bénéfice franc en termes de survie (> 20 %) pour les rechutes de LAL-B pédiatriques de haut risque (13). Son utilisation se développe désormais en alternance avec la chimiothérapie conventionnelle, car, d’une part, son efficacité et sa tolérance sont moindres en cas de charge blastique importante et, d’autre part, son utilisation prolongée conduit à une sélection de blastes n’exprimant pas le CD19. Enfin, sa tolérance est excellente, en particulier hématologique et infectieuse, en faisant un allier précieux dans les situations de toxicité aiguë importante ou de patients très fragiles comme les enfants porteurs de trisomie 21 ou les nourrissons.
La daratumomab
Comme souvent, les LAL-T ont vu un développement moindre d’anticorps thérapeutique et la cible principale reste le CD38, aussi présent sur les LAM, avec le daratumomab. Du fait de son efficacité inconstante, son positionnement est encore à définir, peut-être en association avec le vénétoclax (Fig. 4).
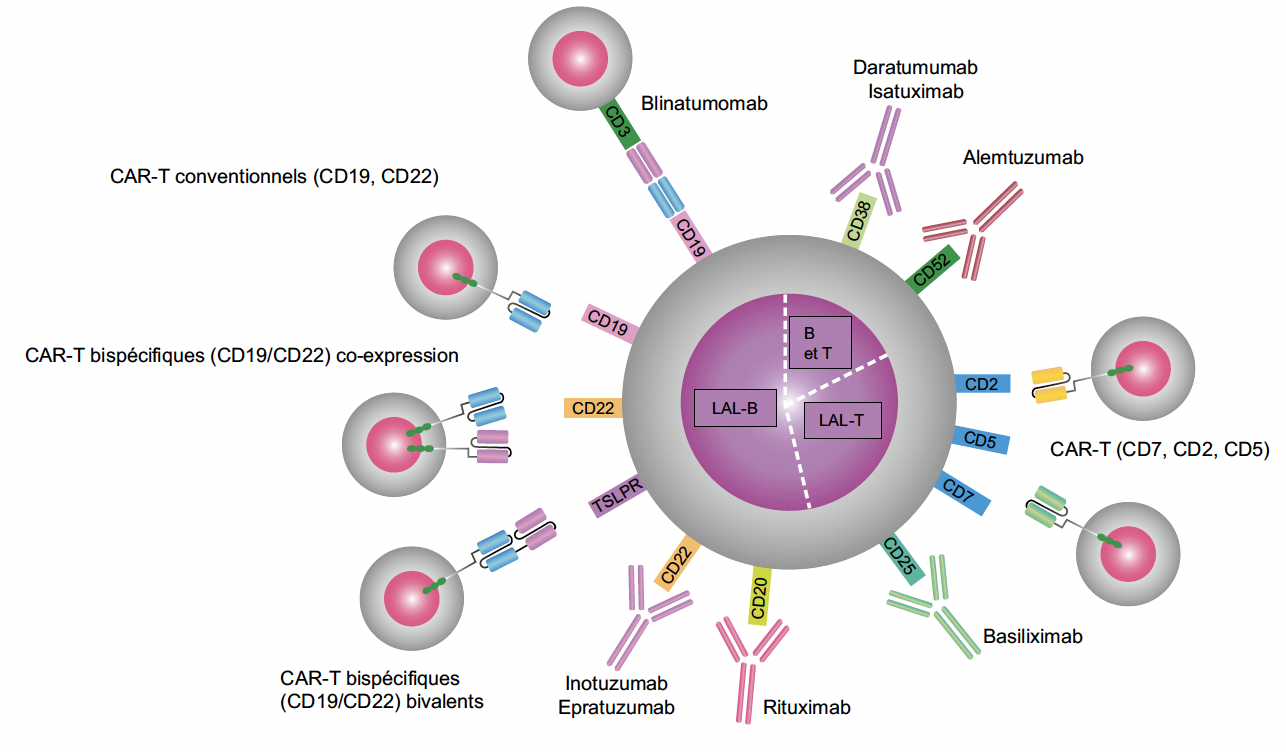
Figure 4 – Immunothérapie dans les LAL (d’après 2).
Le gemtuzumab ozogamicine
Enfin, dans les LAM, le principal anticorps monoclonal utilisé est le gemtuzumab ozogamicine (GO) qui est un anti-CD33 humanisé, couplé lui aussi à la calichéamicine. Il est désormais introduit en association avec la cytarabine et une anthracycline dans les protocoles de phase III européens ou américains en 1re ligne (Myechild01, NCT02724163 et MDCC, NCTT04915612) (3).
De façon récente, un anticorps bispécifique anti-CD123/CD3 est en cours de développement (flotétuzumab) comme aussi des anticorps bispécifiques CD33/CD3.
Les CAR-T cells
Dans les LAL-B
Cette immunothérapie cellulaire adoptive a révolutionné la prise en charge des LAL-B réfractaires. Les CAR-T développés ces dernières années ciblent principalement le CD19 et comprennent dans leur récepteur un domaine de stimulation (CD3) et de costimulation (4-1BB, CD28). Contrairement au blinatumomab, le développement des CAR-T dans la LAL a été plus rapide chez l’enfant que chez l’adulte, en raison d’un risque élevé de neurotoxicité centrale et de syndrome de relargage de cytokine chez les adultes plus âgés. Le tisagenlecleucel (tisa-cel) est la seule formulation à avoir une AMM en 2e rechute ou 1re rechute post-allogreffe chez l’enfant et l’adulte jeune de moins de 26 ans. La perte précoce du CAR-T ou la perte de la cible CD19 par les blastes sont les deux principaux mécanismes d’échappement à l’origine des rechutes post-CAR-T.
Les nouvelles stratégies visent à développer des CAR-T persistants aux toxicités moindres et/ou ciblant un ou plusieurs antigènes B (CD19, CD20, CD22) afin de prévenir ces rechutes (14). Des approches de CAR-T allogéniques sont également étudiées permettant un accès plus rapide à la thérapie. Le développement en 1re ligne se poursuit chez l’enfant avec notamment l’essai international ALL1721/CASSIOPEE (NCT03876769) qui inclut des enfants et adolescents en mauvaise réponse moléculaire initiale.
Dans les LAL-T
Les premiers essais utilisant des CAR-T anti-CD7 et ciblant les LAL-T sont attendus avec intérêt compte tenu du pronostic très défavorable de ces LAL en rechute. Plusieurs méthodes sont proposées pour annihiler l’expression de CD7 à la surface des CAR-T et éviter la lutte fratricide entre cellules T modifiées, mais l’aplasie T induite conduit à la nécessité d’une allogreffe de CSH ultérieure (Fig. 4).
Dans les LAM
Le CD123 et le CD33 sont eux les cibles de choix pour le développement des CAR-T cells dans les LAM. En effet, ils sont présents dans plus de 80 % d’entre elles. La myélotoxicité de CD123 et de CD33 reste un facteur limitant, même si des techniques d’expression transitoire et/ou conditionnelle sont proposées et qu’ils sont utilisés en “pont” prégreffe. Des approches combinant plusieurs cibles sont aussi en phase préclinique.
Les soins de support et le suivi à long terme
L’intensification des traitements chimiothérapiques pour améliorer les résultats des LA à haut risque de rechute a entraîné une morbidité importante et une mortalité chez des patients en rémission. Une maîtrise judicieuse des soins de support est donc primordiale.
Les traitements prophylactiques
Par exemple, plus de la moitié des patients pédiatriques atteints de LAM souffrira d’une infection bactérienne grave et approximativement 10 % expérimenteront une infection fongique invasive. Ainsi, une prophylaxie efficace doit être mise en place pour les patients à haut risque infectieux comme le sont aussi les LAL-T de haut risque. La toxicité des corticoïdes (infections, myopathies, ostéonécroses…), en particulier la dexaméthasone, est aussi mieux connue comme celle de l’asparaginase, médicament devenu central des LAL, dont les formes pégylées à demi-vie longue avec une activité enzymatique vérifiée par des dosages sanguins conduisent à des effets puissants, mais nécessitent une gestion des effets indésirables étroite (pancréatites, thromboses, réactions allergiques…). Le traitement prophylactique ou curatif des atteintes méningées a aussi beaucoup évolué avec une diminution progressive de l’utilisation de l’irradiation neuroméningée au profit des injections intrathécales de chimiothérapie ou de l’utilisation de chimiothérapie systémique à pénétration méningée (méthotrexate à haute dose, dexaméthasone par exemple). En effet, l’irradiation méningée, qu’elle soit à 24, 18 ou 12 Gy entraîne un risque accru de tumeurs secondaires, de retard de croissance, d’endocrinopathies diverses, voire d’effets neurocognitifs.
Le suivi à long terme
De façon générale, la maladie elle-même, la lourdeur des thérapeutiques, les difficultés du parcours exposent à des effets secondaires tardifs, qui peuvent retentir sur l’état de santé, la qualité de vie et l’insertion sociale, longtemps après la fin des traitements. Le suivi à long terme des enfants traités pour leucémie dans l’enfance s’est ainsi organisé depuis 20 ans avec un enjeu double :
• prise en charge individuelle des patients
• et amélioration de nos connaissances.
En France, le programme LEA (pour Leucémie enfants adolescents) est dédié au suivi prolongé après traitement d’une leucémie de l’enfance (15). Le suivi doit être adapté aux facteurs de risque :
• traitements de la leucémie (avec une attention particulière en cas de rechute, irradiation et allogreffe),
• facteurs environnementaux, génétiques et sociaux.
Ce suivi permet une détection précoce des complications comme la cardiomyopathie aux anthracyclines, les cancers secondaires, le syndrome métabolique et ses répercussions cardiovasculaires, les troubles de croissance et l’infertilité et, surtout, leur prise en charge.
Perspectives
• Les avancées futures devront s’appuyer sur des modèles expérimentaux qui reproduiront mieux les leucémies humaines pour innover plus rationnellement et tester la résistance des cellules leucémiques plus efficacement qu’avec les patient-derived xenografts (PDX) en cas de rechute ou de forme réfractaire.
• De plus, l’utilisation plus large d’un monitorage des dosages sanguins des médicaments ou de leur activité (ex. : inhibiteurs de kinase ou asparaginase) devrait aussi améliorer la personnalisation du traitement en prenant en compte les interactions entre le médicament et le patient. Il est en effet important de tenir compte non seulement des caractéristiques des blastes leucémiques, mais aussi de leur environnement au sein du patient.
• Des avancées sont attendues en pharmacogénétique, qui n’est pour l’instant utilisée que pour l’ajustement du 6-mercaptopurine et du méthotrexate, deux composants clés du traitement des LAL. Les différences interindividuelles de réponse aux médicaments concernent, pour autant, bien d’autres molécules et représentent une cause importante de résistance et de toxicité au traitement. L’identification des déterminants pharmacogénétiques des médicaments utilisés pour le traitement des LA permettrait ainsi de repérer les patients répondant de façon sous optimale ou à risque de toxicités aiguës importantes, et d’apporter une personnalisation aux protocoles traditionnels en ajustant la dose des médicaments selon le génotype. Ces informations pourraient aussi affecter les modalités de suivi à long terme en identifiant des profils génomiques exposant ou protégeant le patient des effets indésirables à long terme (par exemple, intérêt potentiel des modalités de surveillance cardiaque après anthracyclines).
• Enfin, l’analyse des variations génétiques germinales permettra aussi de démêler le rôle des polymorphismes héréditaires dans la susceptibilité à développer une leucémie.

Les auteurs déclarent ne pas avoir de liens d’intérêt en rapport avec cet article.
Bibliographie
1. Lacour B, Guyot-Goubin A, Guissou S et al. Incidence of childhood cancer in France: National Children Cancer Registries, 2000-2004. Eur J Cancer Prev 2010 ; 19 : 173-81.
2. Inaba H, Mullighan C. Haematologica 2020 ; 105 : 2524-39.
3. Cooper TD, Alonzo TA, Tasian SK et al. Children’s Oncology Group’s 2023 blueprint for research: myeloid neoplasms. Pediatr Blood Cancer 2023 ; 70 : e30584.
4. Mullighan CG, Goorha S, Radtke I et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007 ; 446 : 758-64.
5. Gu Z, Churchman ML, Roberts KG et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet 2019 ; 51 : 296-307.
6. Tran TH, Hunger SP. The genomic landscape of pediatric acute lymphoblastic leukemia and precision medicine opportunities. Sem Cancer Biol 2022 ; 84 : 144-52.
7. Roberts KG, Mullighan CG. The biology of B-progenitor acute lymphoblastic leukemia. Cold Spring Harb Perspect Med 2020 ; 10 : a034835.
8. Elgarten CW, Aplenc R. Pediatric acute myeloid leukemia: updates on biology, risk stratification and therapy. Curr Opin Pediatr 2020 ; 32 : 57-66.
9. Mullighan CG, Su X, Zhang J et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med 2009 ; 360 : 470-80.
10. Pui CH, Pei D, Raimondi SC et al. Clinical impact of minimal residual disease in children with different subtypes of acute lymphoblastic leukemia treated with Response-Adapted therapy. Leukemia 2017 ; 31 : 333-9.
11. Peters C, Dalle JH, Locatelli F et al. Total body irradiation or chemotherapy conditioning in childhood ALL: a multinational, randomized, noninferiority phase III study. J Clin Oncol 2021 ; 39 : 295-307.
12. Shah RM. Contemporary haploidentical stem cell transplant strategies in children with hematological malignancies. Bone Marrow Transplant 2021 ; 56 : 1518-34.
13. Locatelli F, Zugmaier G, Rizzari C et al. Effect of blinatumomab vs chemotherapy on event-free survival among children with high-risk first-relapse B-cell acute lymphoblastic leukemia. A randomized clinical trial. JAMA 2021 ; 325 : 843-54.
14. Maude SL, Laetsch TW, Buechner J et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018 ; 378 : 439-48.
15. Saultier P, Michel G. How I Treat: long-term survivors of childhood acute leukemia. Blood 2024 ; 143 : 1795-1806.