Comme chaque année, nous vous proposons un tour d’horizon des nouvelles molécules disponibles en oncologie. L’année 2025 a été marquée par une accélération du nombre d’indications remboursées dans le droit commun, mais une diminution du nombre de nouveaux traitements en accès précoce ou compassionnel. La dynamique reste forte du côté des thérapies ciblées et immunothérapies, y compris pour des cancers localisés ou localement avancés. Les informations présentées dans ce dossier ont été rédigées selon les disponibilités en date du 31 décembre 2025.
Résumé
Durant l’année 2025, trois molécules ont été mises à disposition via une autorisation d’accès compassionnel (AAC) et quatre molécules via une autorisation d’accès précoce (AAP). Parmi les molécules déjà disponibles dans le cadre d’AAP ou d’AAC, 14 ont bénéficié d’une autorisation de mise sur le marché (AMM) et d’un remboursement en droit commun.
Abstract
Anticancer treatments available in 2025 in France
During the year 2025, 3 molecules were made available through compassionate access authorization (AAC) and 4 molecules through early access authorization (AAP). Among the molecules already available under AAP or AAC, 14 have been granted marketing authorization (AMM) and reimbursed.
Introduction
Les dispositifs de mise à disposition des médicaments
Depuis la réforme de 2021, deux grands cadres ont été mis en place :
• l’accès précoce (AAP) remplace les ATU de cohorte et concerne les médicaments destinés à être commercialisés dans l’indication concernée. Il est octroyé par la haute autorité de santé (HAS) à la demande du laboratoire pour une durée de 1 an renouvelable. Il peut être demandé avant l’obtention de l’AMM (AAP pré-AMM), mais également après l’obtention de celle-ci (AAP post-AMM), pour les médicaments dont le remboursement n’est pas encore fixé ;
• l’accès compassionnel (AAC) concerne les médicaments non destinés à être commercialisés dans l’indication concernée (pas de démarche en cours en vue d’une AMM) et comprend deux sous-catégories :
- le cadre de prescription compassionnel (CPC, qui remplace les RTU) encadre la prescription d’un médicament non conforme à son AMM. Cette autorisation est octroyée par l’Agence nationale de sécurité du médicament (ANSM) pour une durée de 3 ans renouvelable ;
- l’autorisation d’accès compassionnel (AAC, qui remplace l’ATU nominative) a pour vocation l’accès à un médicament qui ne possède pas d’AMM et dont le laboratoire n’a pas prévu la commercialisation en France. Elle est demandée à l’initiative d’un prescripteur pour un patient donné. La durée d’une AAC est de 1 an renouvelable.
Une étude menée à partir du Système national des données de santé (SNDS) a mis en évidence une croissance importante du nombre de patients traités en ATUc/AAP en oncologie, passant de 3 137 patients en 2019 à 18 341 patients en 2022 (1). Cependant, l’accès effectif à ces traitements reste contraint par la complexité des procédures administratives et de recueil des données (2).
L’ensemble des spécialités disponibles dans le cadre de ces dispositifs sont listées sur le site de l’ANSM (ansm.sante.fr/documents/reference/referentiel-des-specialites-en-acces-derogatoire) ainsi que sur le site OncoAccess (oncoaccess.sfpo.com) qui est régulièrement mis à jour par la Société française de pharmacie oncologique (SFPO). Nous détaillons dans cet article les nouvelles indications disponibles en accès précoce ou compassionnel en 2025 (Tab. 1).
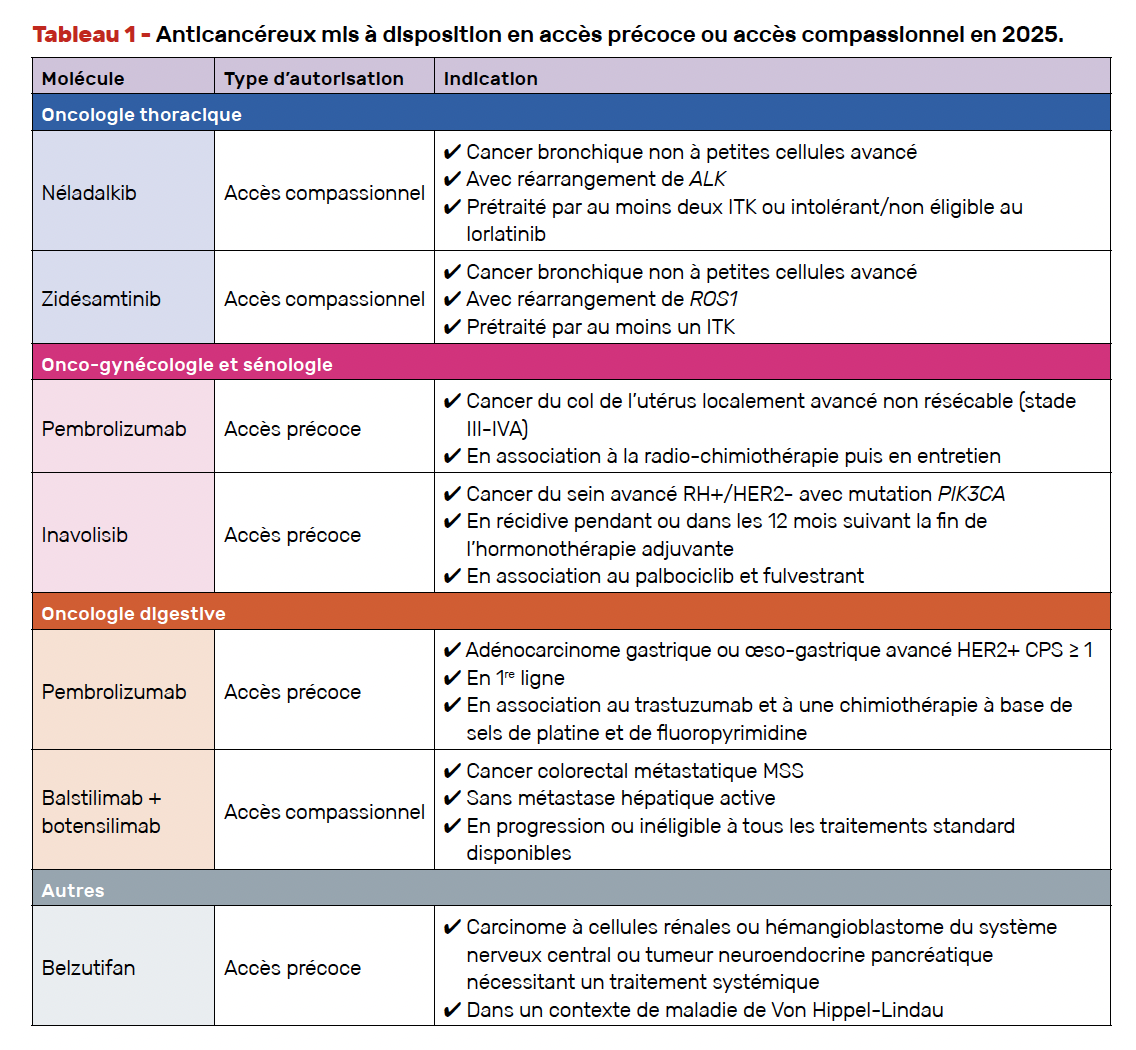
Les accès précoces obtenus en 2025
On notera que certains traitements auparavant disponibles en accès compassionnel sont désormais disponibles en accès précoce, dans l’attente de leur remboursement pérenne.
• Le mirvetuximab soravtansine est disponible en accès précoce pour les patientes atteintes de carcinome séreux de haut grade de l’ovaire, positif au récepteur alpha du folate (FRα), résistant aux sels de platine et ayant reçu une à trois lignes de traitement systémique antérieur.
• Le nirogacestrat est disponible en accès précoce pour les patients adultes présentant une tumeur desmoïde en progression après au moins une ligne de traitement par inhibiteur de tyrosine kinase.
• Le zanidatamab est disponible en accès précoce pour les patients adultes atteints d’un cancer des voies biliaires HER2+ (IHC 3+) avancé, prétraités par gemcitabine et inéligibles au FOLFOX.
Les remboursements obtenus en 2025
Par ailleurs, nous pouvons nous réjouir de nombreux avis favorables de la commission de la transparence de la HAS permettant un remboursement pérenne pour des traitements auparavant disponibles en accès dérogatoire.
• L’alectinib pour le traitement adjuvant après chirurgie R0 des patients adultes atteints d’un cancer bronchique non à petites cellules (CBNPC) ALK+ à haut risque de récidive.
• L’association amivantamab-lazertinib en première ligne de traitement des adultes atteints d’un CBNPC avancé avec mutations de l’EGFR par délétions dans l’exon 19 ou substitution L858R dans l’exon 21.
• L’association dabrafénib-tramétinib sous forme pédiatrique buvable chez les patients pédiatriques âgés d’au moins 1 an atteints d’un gliome avec mutation BRAF V600E (en première ligne dans les gliomes de bas grade et après radiothérapie et/ou chimiothérapie dans les gliomes de haut grade).
• Le dostarlimab en association avec la chimiothérapie par carboplatine paclitaxel en première ligne chez les patientes atteintes d’un cancer de l’endomètre avancé dMMR/MSI-high.
• L’enfortumab védotin en monothérapie dans le carcinome urothélial avancé après chimiothérapie à base de sels de platine et anti-PD-(L)1.
• L’erdafitinib dans le carcinome urothélial métastatique avec altération FGFR3, en progression après chimiothérapie et immunothérapie.
• Le fruquintinib dans le cancer colorectal métastatique après échec des chimiothérapies standard et après trifluridine-tipiracil ou régorafénib.
• L’ivosidenib chez les patients atteints de cholangiocarcinome avancé avec mutation IDH1 R132, prétraités par chimiothérapie et inéligibles au FOLFOX.
• La radiothérapie interne vectorisée par lutécium-177 chez les patients atteints d’un cancer de la prostate métastatique résistant à la castration, exprimant le PSMA et prétraités par hormonothérapie de nouvelle génération ainsi que par chimiothérapie à base de taxane.
• L’association nivolumab-ipilimumab en traitement de première ligne chez des patients adultes atteints d’un cancer colorectal métastatique dMMR/MSI-H.
• Le nivolumab en association à la chimiothérapie à base de sels de platine dans le traitement néoadjuvant des patients atteints de CBNPC résécable à haut risque de récidive, avec expression de PD-L1 ≥ 1 et sans anomalie d’EGFR ni ALK.
• Le sacituzumab govitécan chez les patientes atteintes d’un cancer du sein RH+/HER2- ayant reçu au moins deux lignes de chimiothérapie.
• Le selpercatinib chez les patients atteints d’un cancer médullaire de la thyroïde avancé avec mutation RET en première ou deuxième ligne.
• Le tébentafusp dans le mélanome uvéal avancé chez les patients HLA-A*02:01.
• Le trastuzumab déruxtécan dans le cancer du sein métastatique HER2+ après au moins deux lignes de traitement anti-HER2, ainsi que dans le cancer du sein métastatique HER2 faible après au moins une ligne de chimiothérapie.
Les accès compassionnels perdus en 2025
En revanche, les traitements suivants, auparavant en accès compassionnel, ne sont plus disponibles :
• l’adagrasib dans le traitement des tumeurs solides avancées avec mutation KRAS G12C (y compris CBNPC) ;
• le zongertinib dans les CBNPC avec mutation HER2 en impasse thérapeutique.

Le néladalkib
Le néladalkib (anciennement NVL-655) est un inhibiteur de tyrosine kinase (ITK) sélectif d’ALK doté d’une bonne pénétrance au niveau cérébral et actif sur diverses fusions d’ALK et mutations de résistance. Il épargne par ailleurs la voie TRK, réduisant ainsi les toxicités neurologiques.
L’étude ALKOVE-1
L’essai ALKOVE-1 est une étude de phase I/II visant à étudier l’efficacité et la tolérance du néladalkib chez des patients atteints d’un cancer solide ALK+ et déjà traités par au moins une ligne de traitement systémique (5).
Schéma – Les critères de jugement principaux étaient le taux de réponse objective (TRO), la durée de réponse et la tolérance. L’essai a étudié le néladalkib chez 133 patients (131 avec un CBNPC et deux autres tumeurs, 79 % prétraités par lorlatinib) durant la phase I et la dose de 150 mg a été sélectionnée au vu du profil de tolérance/efficacité.
Résultats – Parmi les 103 patients évaluables pour l’efficacité, le TRO était de 38 %, avec une durée de réponse supérieure à 6 mois chez 79 % d’entre eux. Dans les analyses en sous-groupe, le bénéfice semblait plus important chez les patients naïfs de lorlatinib (53 % de réponse objective) et chez les patients présentant une mutation ALK G1202R (76 %).
Tolérance – Les effets indésirables les plus communs restent l’augmentation des transaminases pour près d’un tiers des patients, de la constipation (15 %), des nausées (12 %) et de la dysgueusie (11 %). Ces toxicités semblent acceptables au vu d’une nécessité d’arrêter le traitement pour seulement 2 % des patients.
Au vu des résultats de cette étude de phase I/II, un accès compassionnel est disponible dans les centres ayant participé à l’essai clinique pour les patients atteints d’un CBNPC ALK+ déjà prétraités par au moins deux ITK ou intolérants/non éligibles au lorlatinib.
Le zidésamtinib
Le zidésamtinib est un ITK sélectif de ROS1 (épargnant la voie TRK), avec une bonne capacité de diffusion intra-cérébrale et actif sur les mutations de résistance telles que G2032R.
L’étude ARROS-1
L’étude ARROS-1 est un essai de phase I/II ayant inclus des patients atteints de cancers solides ROS1+ et déjà prétraités.
Schéma – Au total, 104 patients (dont 99 avec CBNPC) ont reçu le zidesamtinib durant la phase I (escalade de dose de 25 à 150 mg par jour) et 73 d’entre eux ont été inclus dans l’étude d’efficacité de phase II (recevant 100 mg par jour) (6). La plupart des patients avaient reçu au moins trois lignes de traitement systémique, et 99 % d’entre eux au moins un ITK ciblant ROS1.
Résultats – Le TRO était de 38 % pour la population globale et de 45 % chez les patients non exposés au répotrectinib. Chez les patients pré-exposés uniquement au crizotinib, le taux de réponse était de 64 %. Par ailleurs deux patients étaient en réponse complète avec une durée de 16,6 et 23,5 mois respectivement. Pour les patients avec mutation ROS1 G2032R, les résultats restaient satisfaisants avec un TRO de 65 % chez les patients naïfs de répotrectinib et de 38 % pour les patients prétraités par répotrectinib. Les patients atteints de lésions cérébrales semblaient aussi bénéficier du traitement expérimental avec un taux de réponse intra-cérébrale de 57 %.
Tolérance – Aucun effet indésirable n’a entraîné l’arrêt du traitement et seulement 7,7 % étaient considérés comme de grade supérieur ou égal à 3. On note une réduction de dose nécessaire chez 5,8 % des patients. Les effets indésirables les plus communs étaient les œdèmes périphériques (18 %) et l’augmentation des transaminases (12 %).
Le zidésamtinib est disponible en accès compassionnel pour les patients atteints d’un CBNPC cellules ROS+, ayant reçu au moins une ligne de traitement par un ITK de ROS1, pour les centres ayant participé à l’essai clinique ARROS–1.

L’inavolisib
Le pronostic des cancers du sein RH+/HER2- avancés a été largement amélioré par l’ajout des inhibiteurs de CDK4/6 et de PI3K/mTOR à l’hormonothérapie, mais l’apparition de mécanismes de résistance reste une problématique majeure. L’inhibition combinée des voies du récepteur aux œstrogènes, de CDK4/6 et de PI3K avait montré, dans des modèles précliniques, sa capacité à prévenir l’apparition de ces mécanismes de résistance, mais le cumul des toxicités rendait difficile cette approche. L’inavolisib est un inhibiteur sélectif de forme α de la sous-unité catalytique du complexe PI3K, offrant une fenêtre thérapeutique optimisée par rapport aux précédents inhibiteurs de PI3K. Une étude de phase I avait montré un profil de toxicité satisfaisant en association au palbociclib et au fulvestrant, motivant la poursuite de son développement (7).
L’étude INAVO120
Schéma – L’étude INAVO120 est un essai randomisé en double aveugle de phase III, comparant un traitement de première ligne par inavolisib (versus placebo) + palbociclib + fulvestrant chez les patientes atteintes d’un cancer du sein RH+/HER2- avec mutation PIK3CA, localement avancé ou métastatique et ayant rechuté pendant ou dans les 12 mois suivant la fin de l’hormonothérapie adjuvante (les patientes avec un cancer métastatique de novo n’étaient pas incluables) (8). Le critère de jugement principal était la survie sans progression (SSP).
Au total, 325 patientes ont été randomisées, dont 60 % ménopausées, 80 % avec métastases viscérales et 83 % prétraitées par chimiothérapie en situation (néo)adjuvante.
Résultats – La SSP était significativement améliorée avec une médiane passant de 7,3 à 15,0 mois, soit un hazard ratio (HR) à 0,43 (IC 95 % : 0,32-0,59 ; p < 0,001). Le bénéfice était homogène dans l’ensemble des sous-groupes, hormis chez les patients de plus de 65 ans, mais avec un effectif faible. Le TRO était également amélioré, passant de 25,0 à 58,4 %. En revanche, l’analyse intermédiaire n’a pas permis de démontrer d’amélioration statistiquement significative de la survie globale (SG), malgré un HR encourageant à 0,57 (IC 95 % : 0,33-0,99 ; p = 0,03, mais seuil prédéfini à 0,0098).
Tolérance – Le profil de toxicité semble plus favorable que les inhibiteurs de PI3K de première génération, avec tout de même 24 % d’effets indésirables graves et près de 7 % d’arrêts d’au moins un traitement pour toxicité dans le bras expérimental, contre 10,5 et 0,6 % respectivement. Les principales toxicités spécifiques de l’inavolisib étaient la stomatite, les troubles digestifs et l’hyperglycémie.
L’inavolisib est disponible en accès précoce en association avec le palbociclib et le fulvestrant, chez les patients adultes atteints d’un cancer du sein localement avancé ou métastatique RH+/HER2- et présentant une mutation du gène PIK3CA, en récidive pendant ou dans les 12 mois suivant la fin d’une hormonothérapie adjuvante.
Le pembrolizumab
Le traitement du cancer du col de l’utérus localement avancé reposait jusqu’à présent sur la radio-chimiothérapie à base de sels de platine. Le pembrolizumab avait montré son efficacité en situation métastatique, mais n’avait pas encore fait la preuve de son efficacité en situation localement avancée.
L’étude KEYNOTE-A18
Schéma – L’étude KEYNOTE-A18 est une étude de phase III randomisée, en double aveugle, évaluant l’ajout de cinq cycles de pembrolizumab (ou placebo) en association à la chimio-radiothérapie suivis de 15 cycles d’entretien dans les cancers du col localement avancés à haut risque (9). Les critères de jugement principaux étaient la SSP et la SG.
Au total, 1 060 patientes ont été randomisées (1:1), avec un âge médian de 50 ans, une majorité de stades III-IVa, plus de 80 % avec un carcinome épidermoïde et 95 % avec un CPS ≥ 1. La grande majorité (> 90 %) a reçu une radiothérapie d’au moins 70 Gy dose totale (> 90 %).
Résultats – Le pembrolizumab a permis d’augmenter la SSP avec un HR à 0,7 (IC 95 % : 0,55-0,89 ; p = 0,002) et un taux de SSP à 24 mois passant de 57 % dans le groupe placebo à 68 % dans le groupe pembrolizumab. Les résultats apparaissent constants dans les différents sous-groupes testés. L’analyse hiérarchique prévue initialement dans le protocole n’a pas permis de conclure pour le moment sur la SG avec des résultats encore immatures (HR : 0,73 ; IC 95 % : 0,49-1,07).
Tolérance – Les toxicités de grade supérieur ou égal à 3 ont concerné 75 % du groupe pembrolizumab et 69 % du groupe placebo, dont 4 et 1 % de toxicité immuno-médiée respectivement. Les toxicités en lien avec le pembrolizumab ne semblent pas avoir compromis le bon déroulé de la radio-chimiothérapie et le traitement par cisplatine en comparaison avec le groupe placebo.
Le pembrolizumab est disponible en accès précoce en association à la radio-chimiothérapie (radiothérapie externe suivie d’une curiethérapie), dans le traitement des patientes adultes atteintes d’un cancer du col de l’utérus localement avancé de stades III et IVa qui n’ont pas reçu de traitement définitif préalable.

Le pembrolizumab
Le standard de première ligne chez les patients atteints d’un cancer gastrique HER2+ avancé reposait sur une combinaison de chimiothérapie à base de platine et fluoropyrimidine en association au trastuzumab. L’étude KEYNOTE-811 a évalué l’intérêt d’ajouter une immunothérapie par pembrolizumab à ce schéma.
L’étude KEYNOTE-811
L’étude KEYNOTE-811 est une étude de phase III, randomisée, en double aveugle, cherchant à démontrer la supériorité de l’ajout du pembrolizumab à l’association chimiothérapie et trastuzumab par rapport à l’association chimiothérapie et trastuzumab seule, chez les patients atteints d’un adénocarcinome gastrique ou de la jonction œso-gastrique localement avancé non résécable ou métastatique HER2+, en première ligne de traitement (10).
Schéma – Au total, 698 patients ont été randomisés, avec un âge médian de 62 ans, majoritairement masculins (80 %), pour un tiers asiatiques et avec un statut PD-L1+(CPS ≥ 1%) dans plus de 85 % des cas. La chimiothérapie reçue était du CAPOX (capécitabine + oxaliplatine) pour la majorité des patients (85 %) et du 5-FU + cisplatine pour les 15 % restants. Le critère de jugement principal était double, comprenant à la fois la SSP et la SG.
Résultats – La médiane de SSP passait de 8,1 à 10,0 mois avec l’ajout du pembrolizumab, soit une différence modeste de 1,9 mois, mais significative, avec un HR à 0,73 (IC 95 % : 0,61-0,87 ; p = 0,0002). Dans les études en sous-groupe, ce bénéfice n’était pas retrouvé chez les patients avec un CPS < 1. En termes de SG, il n’était pas retrouvé de bénéfice du pembrolizumab dans la population globale avec un HR à 0,84 (IC 95 % : 0,70-1,01). Néanmoins, la SG apparaissait de façon numérique augmentée avec le pembrolizumab dans la population avec un CPS ≥ 1 (HR : 0,81 ; IC 95 % : 0,67-0,98).
Tolérance – L’ajout du pembrolizumab ne semble pas modifier le profil de toxicité, avec 71 % de toxicités de grades ≥ 3 dans le groupe pembrolizumab contre 65 % dans le groupe placebo. L’incidence des effets indésirables graves était identique entre les deux groupes de traitement (46 %).
Le pembrolizumab est disponible en accès précoce en association au trastuzumab et à une chimiothérapie à base de sels de platine et fluoropyrimidine dans le traitement de première ligne des patients atteints d’un adénocarcinome gastrique ou de la jonction œso-gastrique, localement avancé non résécable ou métastatique, HER2+ et exprimant le PD-L1 avec un CPS ≥ 1.
L’association balstilimab + botensilimab
L’immunothérapie a jusqu’à maintenant été décevante dans les cancers colorectaux non-MSI, mais reste un axe de recherche. Le botensilimab est une immunothérapie anti-CTLA4 de nouvelle génération développée en association avec l’anti-PD-1 balstilimab.
L’étude
Schéma – L’association botensilimab + balstilimab a été évaluée dans un essai de phase I avec escalade de dose puis cohorte d’expansion chez des patients atteints de cancer colorectal métastatique MSS prétraités (11). Parmi les 148 patients traités, 101 ont été inclus dans l’analyse d’efficacité avec au moins 6 mois de suivi.
Résultats – Le TRO était de 17 % et le taux de contrôle de la maladie de 61 %, avec une durée médiane de réponse non atteinte et une SSP médiane de 3,5 mois. Dans les analyses en sous-groupes, le bénéfice semblait essentiellement présent chez les patients n’ayant pas de métastases hépatiques “actives”, c’est-à-dire présentes et non traitées.
Tolérance – Le profil de tolérance était cohérent avec ce que l’on peut attendre d’une double immunothérapie, avec 18 % d’interruptions pour toxicité, principalement à type de colites.
Sur la base de ces données très préliminaires, l’association botensilimab + balstilimab est disponible en accès compassionnel pour les patients présentant un cancer colorectal métastatique MSS, sans métastase hépatique active, en progression après tous les traitements standard appropriés et pour lesquels aucune autre option satisfaisante n’est disponible ou recommandée.
Le belzutifan
La maladie de Von Hippel-Lindau est un syndrome génétique rare lié à une mutation germinale du gène VHL et augmentant le risque de tumeurs bénignes, mais également malignes, avec en particulier carcinomes rénaux, tumeurs neuro-endocrines (TNE) pancréatiques, hémangioblastomes du SNC et de la rétine ou encore phéochromocytomes. L’inactivation de VHL induit une accumulation de HIF-2α, entraînant une activation constitutive de la néo-angiogenèse.
Le belzutifan est une petite molécule inhibitrice d’HIF-2α, initialement évaluée dans les carcinomes rénaux à cellules claires sporadiques (12). Au vu de l’activation de cette voie dans les tumeurs liées à la maladie de Von Hippel-Lindau, il apparaissait logique de tester le belzutifan dans cette situation.
L’étude LITESPARK-004
Schéma – L’étude LITESPARK-004 est un essai de phase II monobras évaluant le belzutifan chez les patients atteints de tumeurs solides en lien avec une maladie de Von Hippel-Lindau, localisées, mais nécessitant un traitement systémique de première ligne (13). L’étude comportait plusieurs cohortes : une cohorte de carcinomes à cellules rénales de 61 patients, une cohorte d’hémangioblastomes du SNC de 50 patients et une cohorte de TNE pancréatiques de 22 patients.
Résultats – Le TRO était de 67 % dans les carcinomes à cellules rénales, de 48 % pour les hémangioblastomes et de 91 % pour les TNE pancréatiques, avec 50 % de réponses complètes dans ces derniers. La SSP médiane n’était atteinte dans aucune cohorte.
Tolérance – Le profil de toxicité était principalement marqué par l’anémie et l’hypoxémie.
Le belzutifan est disponible en accès précoce en monothérapie pour le traitement des patients adultes qui nécessitent un traitement pour un carcinome à cellules rénales, des hémangioblastomes du SNC ou des TNE pancréatiques, localisés et associés à la maladie de Von Hippel-Lindau et pour qui les interventions localisées ne sont pas adaptées.
Conclusion
Avec ces nouvelles indications disponibles en 2025, l’arsenal thérapeutique de l’oncologue continue de s’étoffer. De façon rassurante, de nombreuses indications auparavant disponibles en accès dérogatoire ont rejoint le droit commun, assurant un accès pérenne pour nos patients. Cependant, le nombre de nouveaux traitements est inférieur aux années précédentes. Espérons que l’année 2026 apportera son lot d’innovations thérapeutiques pour nos patients.
Les auteurs déclarent ne pas avoir de liens d’intérêt en rapport avec cet article.
Bibliographie
1. Martin T, Rioufol C, Favier B et al. Impact of early access reform on oncology innovation in France: approvals, patients, and costs. BioDrugs Clin Immunother Biopharm Gene Ther 2024 ; 38 : 465‑75.
2. Levêque D, Beck M, Cotton M. Dispensation for access to novel anticancer agents in France. Lancet Oncol 2023 ; 24 : 719‑20.
3. Lu S, Kato T, Dong X et al. Osimertinib after chemoradiotherapy in stage III EGFR -mutated NSCLC. N Engl J Med 2024 ; 391 : 585‑97.
4. Ramalingam SS, Ozguroglu M, Ahn MJ et al. LBA4: Osimertinib (osi) after definitive chemoradiotherapy (CRT) in patients (pts) with unresectable (UR) stage III EGFR-mutated (EGFRm) non-small cell lung cancer (NSCLC): Updated overall survival (OS) analysis from the LAURA study. J Thorac Oncol 2025 ; 20 : S123‑4.
5. Drilon AE, Lin JJ, Johnson ML et al. 1253O Phase I/II ALKOVE-1 study of NVL-655 in ALK-positive (ALK+) solid tumours. Ann Oncol 2024 ; 35 : S802‑3.
6. Besse B, Drilon AE, Cho BC et al. 1256MO Phase I/II ARROS-1 study of zidesamtinib (NVL-520) in ROS1 fusion-positive solid tumours. Ann Oncol 2024 ; 35 : S804‑5.
7. Bedard PL, Jhaveri K, Cervantes A et al. Abstract PD1-02: A phase I/Ib study evaluating GDC-0077 + palbociclib (palbo) + fulvestrant in patients (pts) with PIK3CA-mutant (mut), hormone receptor-positive/HER2-negative metastatic breast cancer (HR+/HER2- mBC). Cancer Res 2021 ; 81 : PD1-02.
8. Turner NC, Im SA, Saura C et al. Inavolisib-based therapy in PIK3CA-mutated advanced breast cancer. N Engl J Med 2024 ; 391 : 1584‑96.
9. Lorusso D, Xiang Y, Hasegawa K et al. Pembrolizumab or placebo with chemoradiotherapy followed by pembrolizumab or placebo for newly diagnosed, high-risk, locally advanced cervical cancer (ENGOT-cx11/GOG-3047/KEYNOTE-A18): a randomised, double-blind, phase 3 clinical trial. Lancet 2024 ; 403 : 1341‑50.
10. Janjigian YY, Kawazoe A, Bai Y et al. Pembrolizumab plus trastuzumab and chemotherapy for HER2-positive gastric or gastro-oesophageal junction adenocarcinoma: interim analyses from the phase 3 KEYNOTE-811 randomised placebo-controlled trial. Lancet 2023 ; 402 : 2197‑208.
11. Bullock AJ, Schlechter BL, Fakih MG et al. Botensilimab plus balstilimab in relapsed/refractory microsatellite stable metastatic colorectal cancer: a phase 1 trial. Nat Med 2024 ; 30 : 2558‑67.
12. Choueiri TK, Bauer TM, Papadopoulos KP et al. Inhibition of hypoxia-inducible factor-2α in renal cell carcinoma with belzutifan: a phase 1 trial and biomarker analysis. Nat Med 2021 ; 27 : 802‑5.
13. Srinivasan R, Iliopoulos O, Beckermann KE et al. Belzutifan for von Hippel-Lindau disease-associated renal cell carcinoma and other neoplasms (LITESPARK-004): 50 months follow-up from a single-arm, phase 2 study. Lancet Oncol 2025 ; 26 : 571‑82.