Introduction
Le myélome multiple (MM) est une hémopathie maligne caractérisée par la prolifération clonale de plasmocytes (cellules terminales de la différenciation lymphocytaire B), responsable d’une infiltration de la moelle osseuse hématopoïétique et dans la majorité des cas de la sécrétion d’une immunoglobuline monoclonale, complète ou incomplète. Il représente 1 à 2 % des cancers et 10 à 12 % des hémopathies malignes, avec environ 5 400 nouveaux cas par an en France. L’âge médian au moment du diagnostic est proche de 70 ans, un tiers des patients ayant plus de 75 ans et un cinquième plus de 80 ans. Il peut également toucher des personnes plus jeunes, puisque près de 3 % des cas sont diagnostiqués avant 40 ans (1). Tous les facteurs de risque du myélome ne sont pas connus à ce jour. Seules les radiations ionisantes liées à des expositions accidentelles et les expositions professionnelles aux pesticides sont des facteurs de risque connus. Il existe de très rares formes familiales.
Dans la plupart des cas, le myélome est une maladie qui tend à devenir chronique, avec la succession de plusieurs phases de rémissions et de rechutes, et la maladie peut le plus souvent être traitée plusieurs fois. Sa prise en charge diagnostique et thérapeutique a connu une évolution rapide au cours des dernières décennies.
Historique
Après les premières descriptions d’une affection associant asthénie, douleurs osseuses et fractures multiples au milieu du XIXe siècle, puis la découverte d’une protéine anormale dans les urines par Henry Bence Jones, Otto Kahler décrit en 1880 la maladie qui portera longtemps son nom.
Les traitements historiques
• Le melphalan, premier traitement efficace, apparaît en 1958 et l’association melphalan prednisone restera un standard de traitement pendant des décennies. Son utilisation à forte dose suivie d’une autogreffe (d’abord de moelle osseuse puis de cellules souches périphériques) apparaît dans les années 1980 et se développe dans les années 1990.
• À ces médicaments historiques se sont ajoutés, à partir des années 2000, les immunomodulateurs (thalidomide, lénalidomide) et les inhibiteurs du protéasome (bortézomib), classes thérapeutiques à l’origine d’une première amélioration significative du pronostic, et qui restent bien présentes dans les schémas de traitement actuels.
Les évolutions thérapeutiques
• Autour de 2015, le développement de nouvelles molécules parmi les inhibiteurs du protéasome (carfilzomib, ixazomib) et les immunomodulateurs (pomalidomide) et surtout l’apparition des anticorps monoclonaux anti-CD38 (daratumumab, isatuximab), dont l’ajout aux schémas traditionnels de traitement a constamment apporté un bénéfice, ont profondément modifié les standards de traitement de rechute, puis de première ligne.
• D’autres molécules au mode d’action original (vénétoclax, séli-nexor, melflufen, anticorps monoclonaux conjugués) ont montré une certaine efficacité dans des essais thérapeutiques.
• Mais c’est surtout l’apparition dans les années 2020 d’immunothérapies innovantes, anticorps bispécifiques et thérapies cellulaires du type Chimeric Antigen Receptor T-Cell (CAR T), qui ont permis d’obtenir des taux et des profondeurs de réponse jamais observés jusqu’alors chez des patients très lourdement prétraités.
Les évolutions des critères diagnostiques et pronostiques
Par ailleurs, les dernières années ont vu s’affiner les critères diagnostiques et pronostiques de la maladie, que ce soit en termes d’imagerie médicale au diagnostic et en cours de suivi (IRM et TEP-scan), d’évaluation pronostique (cytogénétique et génomique) ou d’évaluation de la maladie résiduelle dans la moelle osseuse (analyse génomique et cytométrie en flux) et dans le tissu osseux (TEP-scan). Cette évaluation de la maladie résiduelle (Minimal Residual Disease ou MRD) est devenue pertinente dans le cadre des progrès thérapeutiques évoqués plus haut.
À défaut d’une guérison, qui est désormais l’objectif affiché de la recherche, l’obtention d’une MRD négative permet d’espérer pour de nombreux patients un contrôle prolongé tout en maintenant une qualité de vie correcte.
Physiopathologie : conséquences de la prolifération plasmocytaire
Apparition d’un clone de plasmocytes tumoraux
L’apparition d’un clone de plasmocytes tumoraux puis sa prolifération dans la moelle osseuse sont responsables d’un ensemble d’événements physiopathologiques conduisant aux symptômes classiques du MM regroupés dans l’acronyme CRAB (hyperCalcémie, atteinte Rénale, Anémie, Bone disease (lésion osseuse)) :
1. l’infiltration médullaire par le clone est à l’origine d’une anémie arégénérative, le plus souvent normocytaire ;
2. la sécrétion par les plasmocytes tumoraux de cytokines (IL-6, RANK…) perturbe l’homéostasie du stroma médullaire, en activant les ostéoclastes et en inhibant les ostéoblastes ; ce déséquilibre entre résorption et fabrication osseuse conduit à l’apparition de lésions ostéolytiques, classiquement “à l’emporte-pièce”, principalement situées dans les os riches en moelle : crâne, rachis, côtes, bassin et os longs ;
3. une hypercalcémie par ostéolyse maligne est alors possible ;
4. la production en grande quantité de fragments d’immunoglobuline monoclonale dans les MM dits “à chaînes légères”, qui ne sécrètent pas une immunoglobuline complète, peut conduire à une précipitation dans les tubules rénaux et entraîner une insuffisance rénale, parfois aiguë : c’est la tubulopathie myélomateuse (ou néphropathie à cylindres myélomateux) ; cette précipitation est souvent déclenchée ou aggravée par la déshydratation, l’hypercalcémie, les infections et certains médicaments (anti-inflammatoires non stéroïdiens et produits de contraste iodés en particulier).
Déficit immun humoral profond
D’autre part, la répression concomitante de la synthèse des immuno-
globulines polyclonales est à l’origine d’un déficit immun humoral profond, à l’origine d’une susceptibilité particulière aux germes encapsulés, et notamment au pneumocoque (défaut d’opsonisation permettant une destruction cellulaire efficace).
Diagnostic
Circonstances de découverte
• Les symptômes osseux sont le mode de découverte le plus fréquent, estimé à près de 60 % des cas : douleurs osseuses (et notamment rachidiennes) d’horaire inflammatoire, fractures pathologiques, hypercalcémie.
• L’anémie est également un mode fréquent de découverte, estimé à 30 % des cas.
• La maladie peut se révéler lors de complications infectieuses, notamment à germes encapsulés (pneumonie franche lobaire aiguë, parfois méningite à pneumocoque).
• Le diagnostic est parfois réalisé à l’occasion d’une insuffisance rénale aiguë, éventuellement terminale.
Rappelons enfin que la maladie est découverte dans près d’un tiers des cas en l’absence de tout symptôme, sur un bilan systématique ou réalisé dans un autre cadre.
Diagnostic positif
Le bilan biologique
Si l’examen physique est souvent pauvre et peu informatif, un bilan biologique simple peut orienter :
• hémogramme à la recherche d’une anémie,
• analyse de la fonction rénale,
• dosage de la calcémie,
• électrophorèse des protéines sériques objectivant un “pic” d’allure monoclonale.
Ce pic monoclonal fait défaut dans 10 à 20 % des cas environ, seule la chaîne légère de l’immunoglobuline monoclonale étant produite ; l’électrophorèse ne retrouve alors qu’une hypogammaglobulinémie, d’ailleurs témoin de la répression de synthèse des immunoglobulines polyclonales. La recherche d’une protéinurie de surcharge tubulaire (protéinurie de Bence Jones), constituée de ces mêmes chaînes légères et souvent non détectée à la bandelette, prend alors toute sa valeur.
L’affirmation du caractère monoclonal de la dysglobulinémie et la détermination de son isotype reposent sur l’immunofixation sanguine (et urinaire pour les formes à chaînes légères). Il s’agit le plus souvent d’une IgG pour la chaîne lourde, moins fréquemment d’une IgA (10 à 15 %), très rarement d’une IgD. La chaîne légère est kappa dans deux tiers des cas, lambda pour le dernier tiers.
Le myélogramme
C’est le myélogramme qui permet le diagnostic positif de MM lorsqu’il met en évidence au moins 10 % de plasmocytes dystrophiques.
Diagnostic différentiel
L’augmentation du clone tumoral est progressive et il existe un continuum physiopathologique entre dysglobulinémie monoclonale de signification indéterminée, MM asymptomatique et MM symptomatique.
La dysglobulinémie monoclonale de signification indéterminée
La prévalence des dysglobulinémies monoclonales est loin d’être exceptionnelle à partir d’un certain âge :
• 1 à 2 % dans la tranche des 50-60 ans,
• 3 % dans celle des 60-70 ans,
• 5 % environ dans celle des 70-80 ans
• et pas loin de 10 % ensuite.
La découverte souvent fortuite d’une dysglobulinémie monoclonale de faible taux, en l’absence d’anémie, d’altération de la fonction rénale, de protéinurie de surcharge tubulaire, d’hypercalcémie ou de lésion osseuse, conduit alors à poser le diagnostic de dysglobulinémie monoclonale de signification indéterminée (Monoclonal Gammapathy of Uncertain Significance ou MGUS). Dans ces cas, et notamment lorsque le taux de l’immunoglobuline est très faible et chez les patients très âgés, on peut se dispenser de l’analyse du myélogramme, qui généralement ne retrouverait alors pas d’infiltration plasmocytaire significative.
Le risque de progression d’une MGUS vers un MM est de 1 % par an, très dépendant bien sûr du taux de l’immunoglobuline monoclonale. La surveillance est essentiellement fonction de celui-ci, et également du terrain sous-jacent (âge, comorbidités).
La dysglobulinémie monoclonale de signification clinique
En revanche, il ne faut pas méconnaître la possibilité d’une dysglobulinémie monoclonale dite de signification clinique (Monoclonal Gammopathy of Clinical Significance ou MGCS), où l’immunoglobuline monoclonale en elle-même, même à très faible taux, peut par ses propriétés physicochimiques entraîner des atteintes d’organes parfois sévères : amylose AL (à évoquer devant une macroglossie, des signes d’insuffisance cardiaque, une protéinurie glomérulaire), cryoglobulinémie (purpura vasculaire, douleurs articulaires).
Les lymphomes et leucémies
Rappelons enfin qu’une immunoglobuline monoclonale peut être satellite de certains lymphomes ou d’une leucémie lymphoïde chronique.
Le myélome multiple asymptomatique
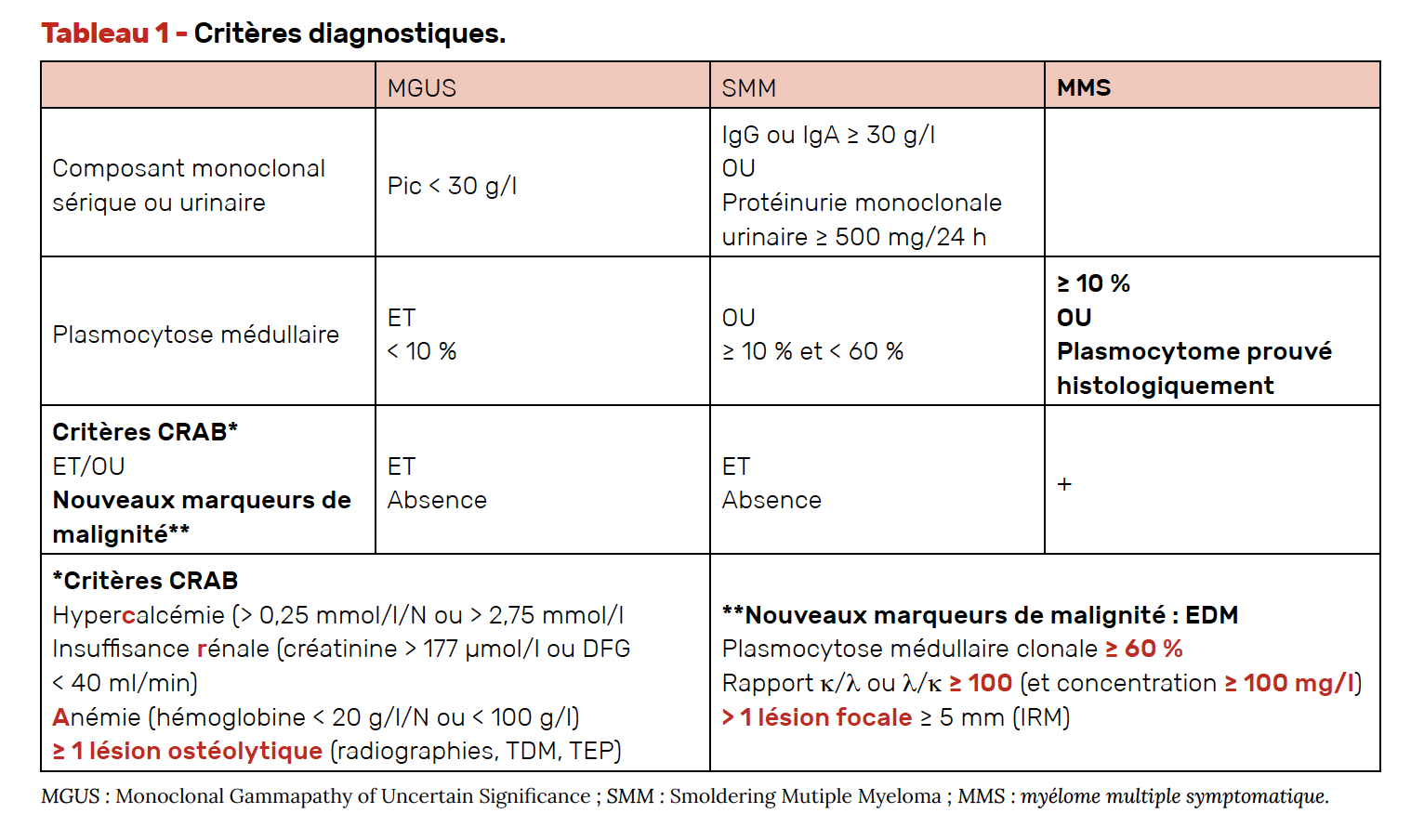
La découverte d’une immunoglobuline monoclonale de taux plus important (≥ 30 g/l) associée à une infiltration plasmocytaire significative au myélogramme (≥ 10 %), mais sans répercussion clinique ou biologique, définit le MM asymptomatique (Smoldering Mutiple Myeloma ou SMM).
À ce jour, en l’absence de traitement curateur et étant donné les différences de profil évolutif, les MM asymptomatiques ne relèvent pas dans la pratique courante d’un traitement antitumoral. Des recherches sont actuellement menées pour discriminer au mieux les formes amenées à évoluer de celles qui resteront indolentes.
Le myélome multiple symptomatique
La définition du MM symptomatique, qui relève d’un traitement spécifique, a été enrichie en 2014 de nouveaux critères sur la base des travaux de l’International Myeloma Working Group (IMWG) (2).
Aux classiques critères CRAB se sont ajoutés des événements définissant le myélome (EDM), à savoir :
1. une infiltration plasmocytaire ≥ 60 % ;
2. et/ou un ratio chaînes légères libres κ/λ ou λ/κ ≥ 100 ;
3. et/ou au moins une lésion focale à l’IRM, d’au moins 5 mm.
Ces nouveaux critères constituent un changement majeur dans la prise en charge du MM, puisqu’ils visent à identifier, parmi les patients sans critères CRAB, ceux avec un risque imminent de progression vers un MM symptomatique et pouvant donc bénéficier d’un traitement précoce avant une atteinte d’organe parfois irréversible (Tab. 1).
Imagerie
Même en l’absence de symptôme, l’atteinte osseuse doit être systématiquement recherchée lorsque le diagnostic positif de MM est retenu.
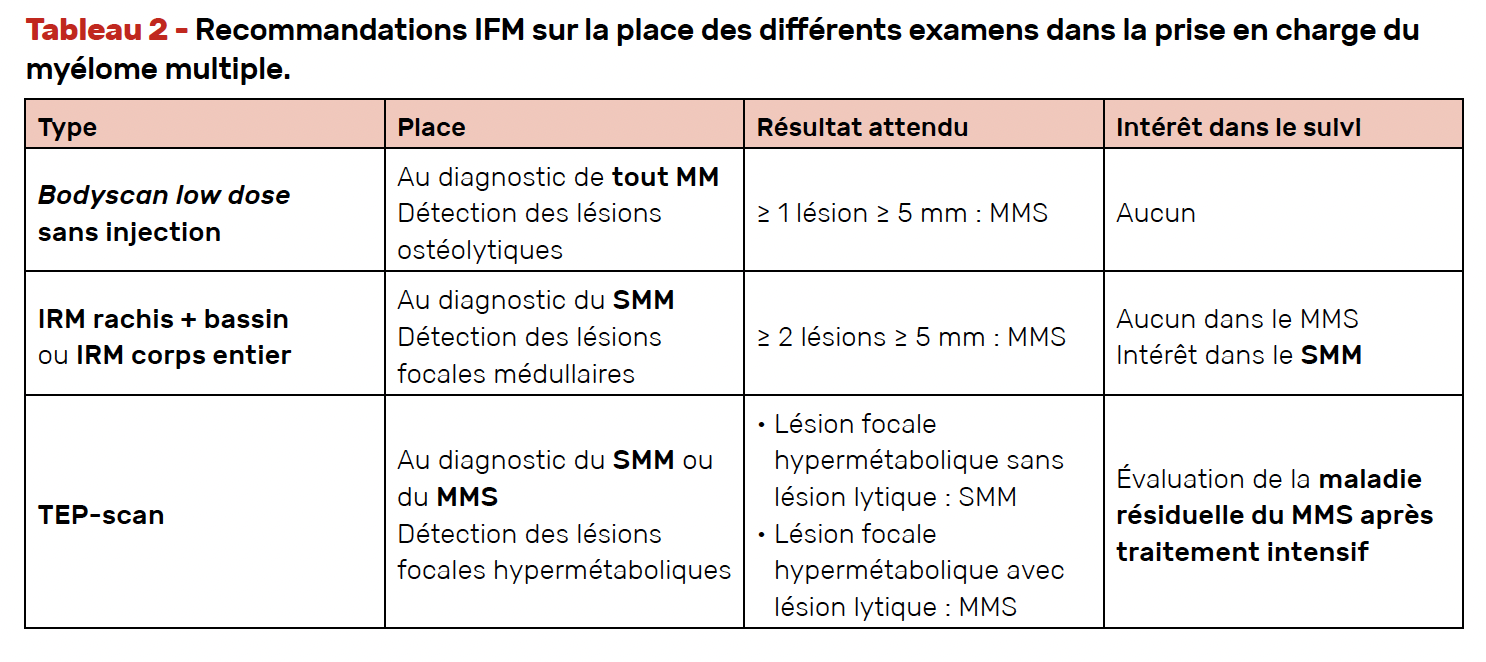
Du fait de leur manque de sensibilité, les clichés radiologiques standard du squelette n’ont plus de place dans le bilan d’extension systématique du MM, qui fait appel selon les indications (et selon leur disponibilité) :
• au scanner osseux low dose corps entier (sans injection),
• à l’IRM du rachis et du bassin (ou mieux à l’IRM corps entier)
• et de plus en plus régulièrement au TEP-scan au 18-FDG.
L’Intergroupe francophone du myélome (IFM) a édité des recommandations sur la place de ces différents examens dans la prise en charge du MM (Tab. 2).
Critères pronostiques
Le MM se distingue par une grande hétérogénéité entre patients, que ce soit dans la présentation clinique initiale ou dans l’évolution de la maladie.
Les classifications à visée pronostique au diagnostic
Les classifications à visée pronostique établies au moment du diagnostic se sont affinées au fil de temps :
1. Classification de Salmon et Durie (1975), reflétant la masse tumorale initiale avec ou sans insuffisance rénale (3) ;
2. International Staging System ou ISS (2005), faisant appel à deux paramètres biologiques de routine (taux d’albumine et de bêta-2-microglobuline) (4) ;
3. Revised International Staging System ou R-ISS (2015), faisant intervenir le taux de LDH et la présence ou non de certaines anomalies cytogénétiques dites de haut risque : del(17p) et/ou t(4;14) et/ou t(14;16), détectées par FISH (5) ;
4. Second Revised International Staging System ou R2-ISS (2022), ajoutant le gain ou l’amplification de 1q aux anomalies cytogénétiques de haut risque déjà prises en compte (6, 7).
Ces trois dernières classifications permettent d’avoir une estimation de la survie globale pour l’ISS, de la survie globale (Overall Survival ou OS) et de la survie sans progression (Progression Free Survival ou PFS) pour les R-ISS et R2-ISS.
Les anomalies cytogénétiques
Concernant les observées dans le MM :
1. leur fréquence est variable : 8 % des patients au diagnostic présentent une délétion 17p [del(17p)] dans plus de 55 % des noyaux analysés, 15 % une translocation (4;14) [t(4;14)], 8 % une délétion (1p32) [del(1p32)], et près d’un tiers un gain ou une amplification de 1q ;
2. la plupart d’entre elles ont une incidence pronostique péjorative, les trisomies 3 et 5 ayant au contraire un effet légèrement favorable ;
3. la t(4;14) a un effet péjoratif hétérogène, aggravé par la présence simultanée d’une del(1p32) ;
4. la del(17p) a un effet constamment péjoratif lorsque la taille du clone délété est ≥ 55 %, et plus encore si elle est associée à une mutation de l’allèle TP53.
Le score pronostique
L’IFM a publié en 2018 un score pronostique en pondérant l’incidence de six anomalies cytogénétiques d’intérêt [trisomie 5, trisomie 21, t(4;14), gain 1q, del(1p32), del(17p)] et prenant en compte des anomalies moléculaires [TP53, NRAS, KRAS, FAM46C, DIS3, ATM, ATR, MYC, TRAF3, BIRC2, BIRC3, CYLD, IRF4, CRBN] permettant de différencier des patients de pronostic favorable, intermédiaire ou défavorable (8).
La négativation de la MRD
Indépendamment du statut cytogénétique initial, la négativation de la MRD dans la moelle osseuse est le plus puissant facteur pronostique de la PFS en cours de traitement ; les patients obtenant cette négativation de la MRD, mesurée dans les essais cliniques par cytométrie de flux ou par séquençage de l’ADN (Next Generation Sequencing ou NGS), ont une meilleure PFS, quel que soit leur risque cytogénétique initial (9).
Un certain nombre d’essais cliniques en cours sont d’ailleurs guidés par la MRD en ce qui concerne l’intensité du traitement.
Stratégie thérapeutique
Moyens thérapeutiques
Les possibilités de traitement du MM ont considérablement évolué au cours des 30 dernières années.
En 2023, l’arsenal thérapeutique comporte en France de nombreuses classes thérapeutiques.
Les corticoïdes : prednisone, dexaméthasone
Ayant une activité antitumorale et antiinflammatoire puissante, synergiques avec l’ensemble des médicaments spécifiques disponibles, ils sont incontournables et font partie de l’ensemble des schémas thérapeutiques.
Les alkylants
Molécules très efficaces sur le plasmocyte tumoral, les alkylants (melphalan, cyclophosphamide) ont encore une place dans certains schémas de première ligne ou de rechute avancée.
Mais c’est surtout l’utilisation de melphalan à dose myélo-ablative, supportée par une autogreffe de cellules souches périphériques, qui reste incontournable dans le traitement de première ligne du sujet éligible à ce type de procédure.
Les inhibiteurs du protéasome : bortézomib, carfilzomib, ixazomib
Les plasmocytes malins sécrètent une grande quantité d’immunoglobulines, nécessitant un turn-over protéique important, ce qui rend les plasmocytes très sensibles à l’inhibition du protéasome.
Les immunomodulateurs (IMIDs) : thalidomide, lénalidomide, pomalidomide
Molécules au mode d’action complexe (propriétés immuno-modulatrices et antiangiogéniques, mais également effet antitumoral direct), les IMIDs sont rapidement devenus incontournables dans une majorité de schémas de traitement.
Les anticorps monoclonaux anti-CD38 : daratumumab, isatuximab
Ils ont ouvert la voie de l’immunothérapie dans le MM. Le récepteur CD38 s’intègre dans des voies de signalisation régulant l’apoptose, la survie et la prolifération des plasmocytes. La fixation de l’anticorps sur le CD38 concourt à la destruction des plasmocytes par induction directe de l’apoptose et activation du système immunitaire (cytotoxicité dépendante du complément, cytotoxicité dépendante de l’anticorps, phagocytose).
D’abord utilisé en monothérapie chez les patients en rechute et réfractaires, avec un taux de réponse avoisinant les 30 %, le daratumumab a vu ses indications remonter les lignes en association à d’autres standards de traitement, initialement en première rechute, puis en première ligne.
Les anticorps bispécifiques : téclistamab, elranatamab
Ils ciblent d’une part le récepteur membranaire CD3 exprimé à la surface des lymphocytes T et d’autre part un antigène de surface plasmocytaire, ici le B-Cell Maturation Antigen ou BCMA. Le rapprochement du lymphocyte T et de sa cible lui permet d’exercer son action cytotoxique.
Les CAR-T cells : idécabtagène-vicleusel
Les lymphocytes T du patient (autologues) sont reprogrammés in vitro par transduction grâce à un lentivirus d’un récepteur chimérique (CAR) qui cible spécifiquement les cellules tumorales, en l’occurrence le BCMA.
Ce système perfectionné permet d’utiliser la propriété de reconnaissance d’un anticorps avec les fonctions performantes de cytotoxicité et de mémoire des lymphocytes T du patient.
Les autres thérapies ciblées
D’autres thérapies ciblées au mode d’action original sont en cours d’essais cliniques ou en attente d’AMM :
• le vénétoclax, inhibiteur spécifique de la protéine antiapoptotique BCL2 ayant un intérêt particulier en cas de translocation t(11;14) ou de niveau élevé d’expression de BCL2 ;
• le sélinexor, inhibiteur sélectif de l’exportine 1 (XPO1).
L’arsenal thérapeutique contre le MM s’est donc considérablement développé, offrant un vaste choix d’options thérapeutiques, que ce soit en première ligne de traitement ou lors des rechutes. Les associations actuellement approuvées et remboursées en France sont nombreuses.
Les soins de support
Par ailleurs, les soins de support sont indispensables à chaque phase de la maladie :
• prise en charge des lésions osseuses : antalgiques, décompression neurochirurgicale de lésions menaçantes, radiothérapie, biphosphonates ;
• prise en charge de l’anémie : support transfusionnel, agents stimulants l’érythropoïèse ;
• prévention ou traitement de l’insuffisance rénale : hydratation, parfois épuration extra-rénale, contre-indication des médicaments néphrotoxiques ;
• prévention des infections : vaccination contre la grippe saisonnière, le SARS-CoV-2, le pneumocoque, immunoglobulines polyvalentes, antibioprophylaxie, notamment contre les réactivations virales et la pneumocystose pulmonaire.
La prise en charge de cette hémopathie maligne, tendant à devenir chronique, se doit d’être pluridisciplinaire.
Stratégie thérapeutique de première ligne (Fig. 1)
Elle fait appel à une distinction entre deux populations :
• celle des patients éligibles à une chimiothérapie intensive avec
autogreffe, avec une limite d’âge classiquement fixée à moins de 66 ans ;
• celle des patients inéligibles à une chimiothérapie intensive avec autogreffe, pour des raisons d’âge ou de comorbidité.
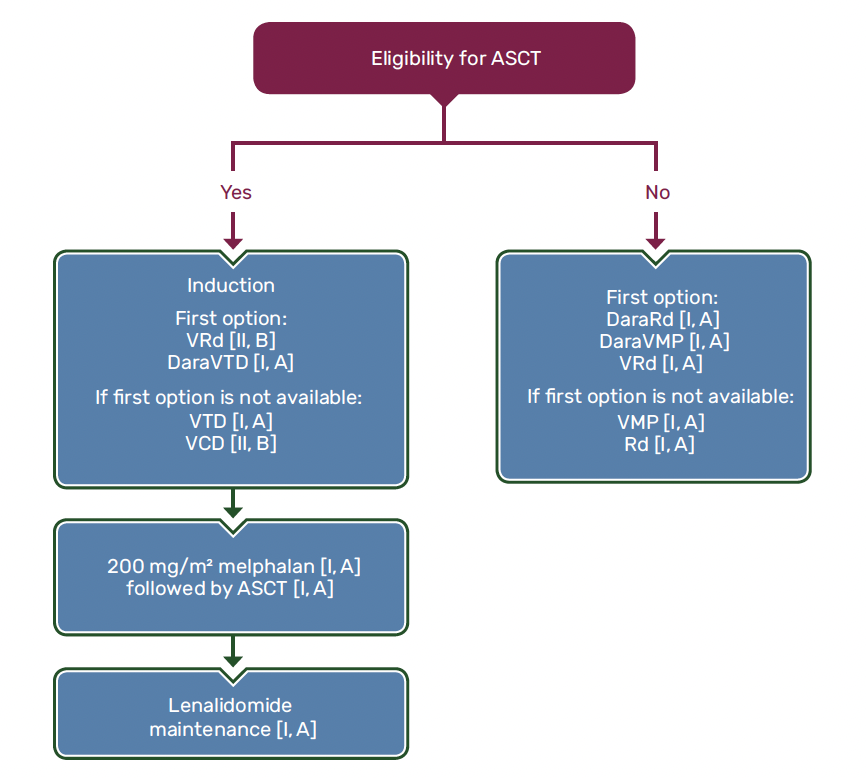
Figure 1 – Recommandations ESMO en première ligne.
La pertinence de ce seuil d’âge, conservée dans les essais cliniques, est discutable du fait de la nette amélioration de l’état général de la population plus âgée, en particulier dans la tranche de 65 à 70 ans.
Sujet éligible à la chimiothérapie intensive avec autogreffe
La chimiothérapie intensive par melphalan à haute dose (200 mg/m²) suivie d’une autogreffe de cellules souches périphériques, dont la supériorité a été démontrée au milieu des années 1990 (10), reste une étape essentielle dans le traitement du MM.
Le traitement se décompose actuellement en quatre phases :
• une phase dite d’induction de 4 mois, comportant désormais une association quadruple (anticorps monoclonal anti-CD38, inhibiteur du protéasome, immunomodulateur et dexaméthasone), qui vise à un premier contrôle de la maladie et de ses symptômes tout en préservant l’hématopoïèse ;
• la phase d’intensification thérapeutique, comprenant l’administration du melphalan 200 mg/m² puis 48 h plus tard l’autogreffe de cellules souches hématopoïétiques, habituellement collectées dans le sang périphérique par cytaphérèse à l’issue du troisième cycle d’induction ; cette autogreffe n’a aucun rôle thérapeutique propre sur le MM, mais représente un traitement de support indispensable permettant de limiter la durée de l’aplasie chimio-induite ; elle nécessite un séjour en secteur stérile et une hospitalisation de 3 à 4 semaines ;
• une phase dite de consolidation de 2 mois, reprenant l’association de la période d’induction ;
• une période dite de maintenance de 3 ans, comportant un traitement immunomodulateur par voie orale à faible dose.
Place de l’intensification avec autogreffe
• L’essai IFM 2009, conduit chez 700 patients de première ligne entre 2010 et 2012 (et donc à l’ère des thérapies ciblées), a objectivé des résultats très clairement en faveur de l’intensification thérapeutique en termes de taux de réponse, de maladie résiduelle, de survie sans progression et de temps moyen jusqu’à nouveau traitement, cohérents dans tous les sous-groupes de patients (11) et confirmés sur le long terme avec un suivi médian de 8 ans. À noter toutefois l’absence de différence statistiquement significative en termes de survie globale (12).
Deux autres essais randomisés à large échelle vont dans le même sens :
• l’essai DETERMINATION, pendant de l’essai IFM 2009 conduit aux États-Unis par l’équipe du Dana Farber Cancer Institute (DFCI), avec pour différence un traitement de maintenance jusqu’à progression (13) ;
• l’essai FORTE, comparant trois bras de traitement : carfilzomib lénalidomide dexaméthasone (KRd) et intensification, KRd 12 cycles, carfilzomib cyclophosphamide dexaméthasone (KCd) et intensification ; les résultats en termes de PFS, de MRD négative et de MRD négative soutenue sont très nettement en faveur du bras KRd et intensification ; on peut au passage souligner que l’intensification ne remplace pas la synergie d’action du carfilzomib avec le lénalidomide puisque des trois bras, le bras KCd et intensification est le plus mauvais (14).
L’intensification thérapeutique garde donc toute sa place en première ligne de traitement en MM en 2023, d’autant plus qu’à l’exception du traitement par CAR-T dans les phases avancées, c’est le seul schéma de traitement à durée fixe dans le MM ; elle permet dans la grande majorité des cas une rémission sans traitement pour une durée variable, de quelques mois à plusieurs années.
Place des quadruplets comportant un anticorps monoclonal anti-CD38
• Elle est devenue la référence, notamment depuis la publication des premiers résultats de l’essai CASSIOPEIA, qui a montré une supériorité nette du bras daratumumab bortézomib thalidomide dexaméthasone (DVTD) sur le bras bortézomib thalidomide dexaméthasone (VTD), que ce soit en termes de profondeur de réponse, de maladie résiduelle négative ou de PFS (15).
• Il en va de même de l’essai du groupe coopérateur allemand GMMG-HD7 comparant une induction isatuximab bortézomib lénalidomide dexaméthasone (Isa-VRD) à une induction bortézomib lénalidomide dexaméthasone (VRD) (16).
Dans cette association quadruple, et bien que ne disposant pas d’une autorisation de mise sur le marché en France dans ce cadre, le lénalidomide tend à supplanter le thalidomide, beaucoup plus toxique.
Effet de la négativation de la MRD en 2023
L’importance de l’obtention d’une MRD négative sur le risque de progression a été bien mise en évidence sur la cohorte de l’essai IFM 2009, montrant non seulement une amélioration de la PFS dans chacun des deux bras de traitement, dans chaque risque cytogénétique, dans chaque stade ISS, mais également une répercussion en termes de survie globale (9).
La négativation de la MRD est donc devenue un surrogate marker de la survie globale en vue d’accélérer l’approbation des nouveaux schémas thérapeutiques.
Sujet inéligible à la chimiothérapie intensive avec autogreffe
Le traitement est alors d’emblée utilisé jusqu’à progression ou toxicité inacceptable ; il comporte désormais systématiquement un anticorps monoclonal anti-CD38, associé à un immunomodulateur ou à une association inhibiteur du protéasome/alkylant et à la dexaméthasone.
Le standard de traitement est représenté par l’association daratumumab lénalidomide dexaméthasone (DRd) selon les données de l’essai MAIA, dont l’actualisation a été présentée à l’ASH 2022 avec un suivi médian de 64,5 mois : la médiane de survie sans progression est de 61,9 mois, la médiane de survie globale n’étant pas atteinte (17, 18).
L’association daratumumab bortézomib melphalan prednisone (DVMP) peut être également utilisée, selon les données de l’essai ALCYONE (19).
Stratégie thérapeutique de première rechute (Fig. 2)
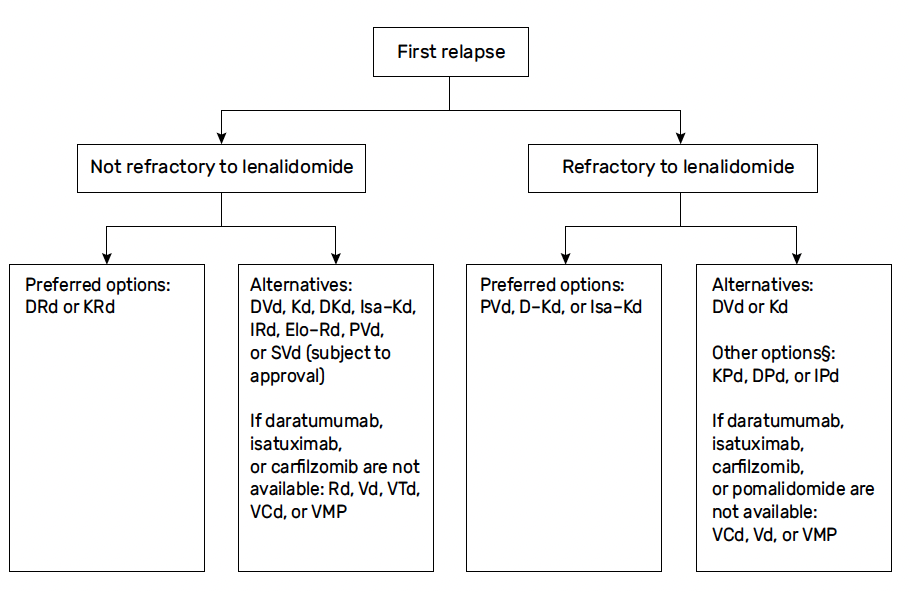
Figure 2 – Recommandations IMWG pour la première rechute.
Malgré les progrès majeurs réalisés au cours des dernières années et les perspectives des traitements de première ligne qu’on présentera plus bas, l’évolution du myélome reste rythmée par la survenue de rechutes successives.
Des taux d’attrition élevés
Chaque nouvelle ligne de traitement est associée à des taux de réponse plus faibles, à une durée de traitement et à des intervalles sans traitement plus courts, à des taux accrus de toxicités et à davantage de comorbidités. Cela est particulièrement vrai chez les sujets âgés, ou qui n’ont pas bénéficié d’une intensification thérapeutique en première ligne.
En vie réelle, il est important d’avoir présent à l’esprit qu’à chaque rechute, entre 15 et 35 % des patients ne reçoivent pas de nouvelle ligne de traitement, et qu’un peu moins de 40 % des patients seulement bénéficient d’une troisième ligne (20).
Ces taux d’attrition élevés soulignent la nécessité d’utiliser les schémas thérapeutiques les plus efficaces, bien sûr en première ligne, mais également dès la première rechute, plutôt que de les réserver pour des lignes ultérieures, où leur bénéfice clinique est plus incertain.
Choix thérapeutiques
En situation de rechute, les choix thérapeutiques dépendent à la fois :
• des caractéristiques du patient : âge, état général, comorbidités, réserves médullaires ;
• de la présentation de la rechute : purement biologique ou clinique, rapidité de progression et agressivité, atteinte extramédullaire ;
• des molécules reçues dans les lignes antérieures ;
• du niveau de réponse, de la durée de réponse et de la tolérance de ces traitements antérieurs ;
• de l’accessibilité de l’association envisagée (21).
Dans les algorithmes proposés par l’International Myeloma Working Group (IMWG) et par l’European Society for Medical Oncology (ESMO), le statut réfractaire ou non au lénalidomide est décisionnel lors de la première rechute (22, 23).
Pour une maladie sensible au lénalidomide
Il est logique de recourir en priorité à une association triple fondée sur le socle lénalidomide dexaméthasone : DRd si cette rechute n’est pas réfractaire aux anticorps monoclonaux anti-CD38, KRd si c’est le cas.
Chaque fois que c’est possible, l’association DRd est utilisée en raison de ses résultats en termes d’efficacité, de sa relative commodité d’utilisation et de son profil de tolérance.
Les résultats à long terme de l’essai POLLUX, avec un suivi médian de 51,3 mois, et pour l’ensemble de la cohorte :
• la PFS dans le bras DRd atteint 45,8 contre 17,5 mois dans le bras Rd ;
• chez les patients en première rechute, elle atteint 53,3 mois ;
• elle reste de 38,8 mois chez les patients exposés mais non réfractaires au lénalidomide.
On note également un bénéfice en termes de PFS2 (53,3 versus 31,6 mois) (24). Elle est comme toujours encore améliorée en cas d’obtention d’une MRD négative, particulièrement lorsqu’elle se maintient plus de 6 mois, voire plus de 12 mois (25).
La survenue d’une rechute réfractaire au lénalidomide présente un caractère défavorable, et ce même avec le recours à des associations n’utilisant pas cette molécule.
Pour une maladie réfractaire au lénalidomide et sensible aux anticorps monoclonaux anti-CD38
On recourra préférentiellement à l’association daratumumab ou isatuximab carfilzomib dexaméthasone (DKd ou Isa-Kd), voire à l’association daratumumab ou isatuximab pomalidomide dexaméthasone (DPd ou Isa-Pd).
Pour une maladie double réfractaire
Les choix sont plus limités, la seule association disponible en France étant l’association bortézomib pomalidomide dexaméthasone (PVd), même si d’autres associations sembleraient plus intéressantes, carfilzomib pomalidomide dexaméthasone (KPd) notamment.
Stratégie thérapeutique des rechutes ultérieures (Fig. 3)
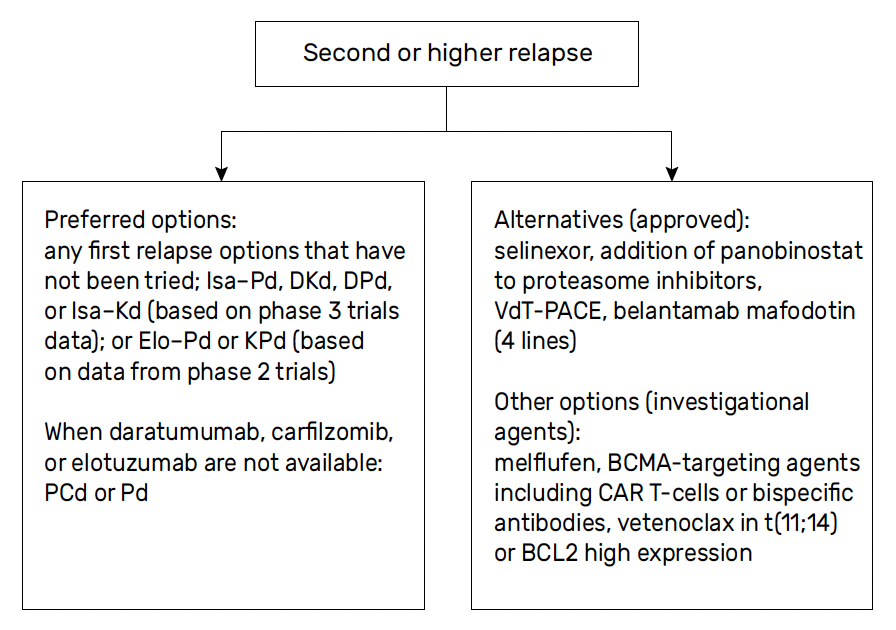
Figure 3 – Recommandations IMWG pour les rechutes ultérieures.
Démarche générale
Dans les algorithmes proposés par l’IMWG et par l’ESMO, il est recommandé de recourir à diverses associations de molécules auxquelles le patient n´a pas été exposé, ou au moins auxquelles il n’est pas réfractaire (22, 23).
La prise en charge est moins standardisée et fait l’objet de discussions parfois complexes en réunion de concertation pluridisciplinaire.
Patients triple exposés et penta-réfractaires
Les possibilités thérapeutiques étaient jusqu’à présent très limitées et l’espérance de survie réduite à quelques mois chez les patients en rechute et réfractaires (26), et notamment chez :
• chez les patients triple exposés, c’est-à-dire exposés aux trois principales classes de médicaments actifs (inhibiteurs du protéasome, IMIDs et anticorps monoclonaux anti-CD38) ;
• a fortiori chez les patients penta-réfractaires, c’est-à-dire réfractaires aux cinq médicaments les plus actifs (bortézomib, carfilzomib, lénalidomide, pomalidomide et un anticorps monoclonal anti-CD38).
Dans ce contexte, les immunothérapies innovantes, anticorps bispécifiques et CAR-T, ont permis d’obtenir des taux et des profondeurs de réponse jamais observés jusqu’alors.
Les anticorps bispécifiques
Les essais de phase II (MajesTEC-1 pour le teclistamab (27), MagnetisMM-3 pour l’elranatamab (28)) ont montré des résultats très encourageants, avec des taux de réponse globale de l’ordre de 60 % chez des patients avec une médiane de cinq lignes antérieures de traitement.
Ces deux molécules sont actuellement disponibles dans le cadre d’un accès précoce.
Un autre anticorps bispécifique ciblant le G protein-coupled receptor, class C group 5 member D ou GPRC5D, le talquétamab, a également montré dans l’essai MonumenTAL-1 un taux de réponse globale proche de 70 % chez des patients triple exposés et penta-réfractaires (29).
CAR-T cells
Les deux CAR-T cells les plus avancées dans les essais cliniques actuellement sont le ciltacabtagène-autoleucel (essai CARTITUDE-1) (30) et l’idécabtagène-vicleusel (essai KarMMa-2) (31).
Ces essais montrent des résultats spectaculaires, avec une mise en rémission complète d’une partie des patients réfractaires à toutes les thérapies disponibles jusque-là. Ainsi, dans l’essai de phase I CARTITUDE-1, le taux de réponse globale atteignait 98 %, avec 80 % des patients en réponse complète stringente.
Ces thérapies présentent cependant des limites : longue durée de fabrication, absence de persistance des CAR-T cells dans l’organisme, difficulté à obtenir une réponse stable dans le temps, avec l’apparition de rechutes plus ou moins précoces.
Perspectives
Perspectives
En première ligne de traitement chez le sujet éligible à la chimiothérapie intensive avec autogreffe
Les essais cliniques en cours évaluent :
• des stratégies de traitement guidées par la MRD : essai IFM 2020-02 « MIDAS » [NCT04934475], essai MASTER (32) ;
• la place des immunothérapies innovantes : essai MASTER II [NCT05231629] pour les anticorps bispécifiques, essai CARTITUDE-6 [NCT05257083] pour les CAR-T.
En première ligne de traitement chez le sujet inéligible à la chimiothérapie intensive avec autogreffe
En vue d´améliorer encore les résultats de l’essai MAIA, les essais cliniques en cours évaluent :
• chez le sujet fit, les possibilités d’amélioration de la réponse par l’utilisation de quadruplets : essais IFM 2020-05 BENEFIT [NCT04751877], IMROZ [NCT03319667] et CEPHEUS [NCT03652064] ;
• chez le sujet fit, les possibilités d’amélioration de la réponse par l’utilisation d’un anticorps bispécifique : essai MajesTEC-7 [NCT05552222] ;
• chez le sujet unfit ou frail, les possibilités d’amélioration de la tolérance par un régime d’épargne de corticoïdes : essai IFM 2017-03 [NCT03993912].
En première rechute
Pour les raisons qu’on a vues précédemment : taux d’attrition, caractère défavorable des rechutes réfractaires au lénalidomide, limitation des choix dans les maladies doubles réfractaires… il est nécessaire d’envisager de nouvelles stratégies thérapeutiques, et ce, dès la première rechute.
Il est logique de faire appel aux immunothérapies innovantes actuellement utilisées en phase avancée.
• Un certain nombre d’essais cliniques évaluent l’intérêt en première rechute d´un anticorps bispécifique : essais MajesTEC-3 [NCT05083169], MagnetisMM-5 [NCT05020236] et MonumenTAL-3 [NCT05455320].
• D’autres ont déjà montré des résultats positifs avec les CAR-T : cohorte 2a de l’essai KarMMa-2 (33), essai CARTITUDE-4 (34).

L’auteur déclare avoir des liens d’intérêt avec Amgen, BMS/Celgene, GSK, Janssen, Sanofi, Stemline, Takeda.
Bibliographie
1. Institut National du Cancer, 2018.
2. Rajkumar SV, Dimopoulos MA, Palumbo A et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014 ; 15 : e538-48.
3. Durie BGM, Salmon SE. A clinical staging system for multiple myeloma: Correlation of measured myeloma cell mass with presenting clinical features, response to treatment, and survival. Cancer 1975 ; 36 : 842-54.
4. Greipp PR, San Miguel J, Durie BG et al. International staging system for multiple myeloma. J Clin Oncol 2005 20 ; 23 : 3412-20.
5. Palumbo A, Avet-Loiseau H, Oliva S et al. Revised international staging system for multiple myeloma: a report from International Myeloma Working Group. J Clin Oncol 2015 ; 33 : 2863-9.
6. Palumbo A, Avet-Loiseau H, Oliva S et al. Revised international staging system for multiple myeloma: a report from international myeloma working group. J Clin Oncol 2015 ; 33 : 2863-9.
7. D’Agostino M, Cairns DA, Lahuerta JJ et al. Second revision of the international staging system (R2-ISS) for overall survival in multiple myeloma: A European Myeloma Network (EMN) report within the HARMONY project. J Clin Oncol 2022 ; 40 : 3406-18.
8. Perrot A, Lauwers-Cances V, Tournay E et al. Development and validation of a cytogenetic prognostic index predicting survival in multiple myeloma. J Clin Oncol 2019 ; 37 : 1657-65.
9. Perrot A, Lauwers-Cances V, Corre J et al. Minimal residual disease negativity using deep sequencing is a major prognostic factor in multiple myeloma. Blood 2018 ; 132 : 2456-64.
10. Attal M, Harousseau JL, Stoppa AM et al. A prospective, randomized trial of autologous bone marrow transplantation and chemotherapy in multiple myeloma. Intergroupe Français du Myélome. N Engl J Med 1996 ; 335 : 91-7.
11. Attal M, Lauwers-Cances V, Hulin C et al. IFM 2009 study. Lenalidomide, bortezomib, and dexamethasone with transplantation for myeloma. N Engl J Med 2017 ; 376 : 1311-20.
12. Perrot A, Lauwers-Cances V, Cazaubiel T et al. Early versus late autologous stem cell transplant in newly diagnosed multiple myeloma: long-term follow-up analysis of the IFM 2009 trial. Blood 2020 ; 136 : 39.
13. Richardson PG, Jacobus SJ, Weller EA et al. Triplet therapy, transplantation, and maintenance until progression in myeloma N Engl J Med 2022 ; 387 :132-47.
14. Gay F, Musto P, Rota-Scalabrini D et al. Carfilzomib with cyclophosphamide and dexamethasone or lenalidomide and dexamethasone plus autologous transplantation or carfilzomib plus lenalidomide and dexamethasone, followed by maintenance with carfilzomib plus lenalidomide or lenalidomide alone for patients with newly diagnosed multiple myeloma (FORTE): a randomised, open-label, phase 2 trial. Lancet Oncol 2021 ; 22 : 1705-20.
15. Moreau P, Attal M, Hulin C et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet 2019 ; 394 : 29-38.
16. Goldschmidt H, Mai EK, Bertsch U et al. Addition of isatuximab to lenalidomide, bortezomib, and dexamethasone as induction therapy for newly diagnosed, transplantation-eligible patients with multiple myeloma (GMMG-HD7): part 1 of an open-label, multicentre, randomised, active-controlled, phase 3 trial. Lancet Haematol 2022 ; 9 : e810-21.
17. Facon T, Kumar S, Plesner T et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med 2019 ; 380 : 2104-15.
18. Kumar S. Updated efficacy analysis from the phase III MAIA study in newly-diagnosed multiple myeloma (Abstracts #4553 and #3245). Presented at the 2022 American Society of Hematology Annual Meeting ; 10 décembre 2022; Nouvelle Orléans, Louisiane.
19. Mateos MV, Cavo M, Blade J et al. Overall survival with daratumumab, bortezomib, melphalan, and prednisone in newly diagnosed multiple myeloma (ALCYONE): a randomised, open-label, phase 3 trial. Lancet 2020 ; 395 : 132-41.
20. Fonseca R, Usmani SZ, Mehra M et al. Frontline treatment patterns and attrition rates by subsequent lines of therapy in patients with newly diagnosed multiple myeloma. BMC Cancer 2020 ; 20 : 1087.
21. Moreau P, San Miguel J, Sonneveld P et al. Multiple myeloma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2017 ; 28 : iv52-61.
22. Dimopoulos MA, Moreau P, Terpos E et al. Multiple myeloma: EHA-ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol 2021 ; 32 : 309-22.
23. Moreau P, Kumar SK, San Miguel J et al. Treatment of relapsed and refractory multiple myeloma: recommendations from the International Myeloma Working Group. Lancet Oncol 2021 ; 22 : e105-18.
24. Bahlis NJ, Dimopoulos MA, White DJ et al. Daratumumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: extended follow-up of POLLUX, a randomized, open-label, phase 3 study. Leukemia 2020 ; 34 : 1875-84.
25. Avet-Loiseau H, San-Miguel J, Casneuf T et al. Evaluation of sustained minimal residual disease negativity with daratumumab-combination regimens in relapsed and/or refractory multiple myeloma: analysis of POLLUX and CASTOR. J Clin Oncol 2021 ; 39 : 1139-49.
26. Mateos MV, Weisel K, De Stefano V et al. LocoMMotion: a prospective, non-interventional, multinational study of real-life current standards of care in patients with relapsed and/or refractory multiple myeloma. Leukemia 2022 ; 36 : 1371-6.
27. Moreau P, Garfall AL, van de Donk NWCJ et al. Teclistamab in relapsed or refractory multiple myeloma. N Engl J Med 2022 ; 387 : 495-505.
28. Lesokhin AM, Tomasson MH, Arnulf B et al. Elranatamab in relapsed or refractory multiple myeloma: phase 2 MagnetisMM-3 trial results. Nat Med 2023 ; 29 : 2259-67.
29. Chari A, Minnema MC, Berdeja JG et al. Talquetamab, a T-cell-redirecting GPRC5D bispecific antibody for multiple myeloma. N Engl J Med 2022 ; 387 : 2232-44.
30. Martin T, Usmani SZ, Berdeja JG et al. Ciltacabtagene autoleucel, an anti-B-cell maturation antigen chimeric antigen receptor T-cell therapy, for relapsed/refractory multiple myeloma: CARTITUDE-1 2-year follow-up. J Clin Oncol 2023 ; 41 : 1265-74.
31. Munshi NC, Anderson LD Jr, Shah N et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N Engl J Med 2021 ; 384 : 705-16.
32. Costa LJ, Chhabra S, Medvedova E et al. Daratumumab, carfilzomib, lenalidomide, and dexamethasone with minimal residual disease response-adapted therapy in newly diagnosed multiple myeloma. J Clin Oncol 2022 ; 40 : 2901-2.
33. Usmani S, Patel K, Hari P et al. KarMMa-2 cohort 2a: efficacy and safety of idecabtagene vicleucel in clinical high-risk multiple myeloma patients with early relapse after frontline autologous stem cell transplantation. Abstract #361. Presented at the 2022 American Society of Hematology Annual Meeting. ; 10 décembre 2022; Nouvelle Orléans, Louisiane.
34. San-Miguel J, Dhakal B, Yong K et al. Cilta-cel or Standard Care in Lenalidomide-Refractory Multiple Myeloma. N Engl J Med 2023 ; 389 : 335-47.