Introduction
Les mélanocytes et la choroïde
Le mélanome uvéal (MU), appelé également mélanome de la choroïde, est la tumeur maligne intra-oculaire primaire la plus fréquente chez l’adulte. Il provient des mélanocytes présents dans la couche choroïde de l’œil et représente un peu moins de 5 % de l’ensemble des mélanomes (1). L’origine embryo-logique des mélanocytes uvéaux (issus de la crête neurale antérieure), le microenvironnement tumoral et immunitaire n’ont aucune similitude avec les tissus cutanés, distinguant cette population de tumeurs comme une entité à part entière. Leur oncogenèse, pronostic, sensibilité aux traitements et évolution métastatique sont très différents des mélanomes cutanés (2). Pour rappel, la choroïde est une fine couche de tissu formant la couche intermédiaire de la paroi de l’œil, entre la sclérotique et la rétine. Avec le corps ciliaire et l’iris, la choroïde forme le tractus uvéal. La production de mélanine par les mélanocytes uvéaux est utile pour protéger l’œil des rayonnements ultraviolets. Dans la grande majorité des cas, le mélanome apparaît au niveau de la choroïde. Plus rarement, il peut aussi se localiser sur le corps ciliaire ou l’iris, associé à un meilleur pronostic.
Épidémiologie
En France, 500 à 600 nouveaux cas de mélanomes uvéaux sont diagnostiqués chaque année. L’âge moyen est de 56 ans. La physiopathologie tumorale est encore mal connue, et apparaît très différente de celle du mélanome cutané (3, 4). Il existe un réseau national de prise en charge de ces mélanomes, baptisé MELACHONAT, dont le centre coordinateur est l’Institut Curie à Paris.
Alors que seulement 4 % des patients présentent des métastases détectables au moment du diagnostic, environ 50 % d’entre eux finiront par en développer, majoritairement hépatiques. Le pronostic de cette maladie est très péjoratif, avec jusqu’à récemment une survie médiane de 12 mois, soit moins de 10 % de survie à 2 ans (5-7). De nouveaux traitements sont en train de voir le jour pour le MU, et représentent une réelle avancée pour les patients.
Facteurs de risque
Certains facteurs de risque ont été identifiés grâce à l’étude d’un grand registre américain collectant des données épidémiologiques SEER de 1973 à 2013 (7).
• Le sex-ratio est similaire de même que la latéralité.
• La population caucasienne est largement plus exposée avec 98 % des cas.
• Les individus aux yeux clairs ont deux à trois fois plus de risque d’être atteints que les yeux bruns.
• Il ne semble pas y avoir de lien entre exposition au soleil et apparition de ce mélanome (8, 9).
Dans le cas d’une lésion pigmentaire bénigne (nævus choroïdien), une surveillance régulière est préconisée du fait d’un risque de transformation.
Les autres facteurs de risque, plus rares, sembleraient associés tels que :
• la mélanose acquise primitive (affection de la conjonctive),
• la mélanose oculo-cutanée ou nævus d’Ota (une pigmentation unilatérale congénitale),
• le syndrome du nævus dysplasique, trouble héréditaire appelé mélanomes atypiques multiples familiaux,
• les lits de bronzage et lampes solaires à rayons ultraviolets (UV),
• les professions de soudeurs,
• les syndromes d’immunodéficience acquise (Sida).
Parmi les facteurs de risque héréditaires, il existe une prédisposition génétique familiale avec mutation constitutionnelle de BAP1, qui associe une prédisposition aux mélanomes uvéaux, mésothéliomes malins de la plèvre et carcinomes rénaux (10).
Symptômes et diagnostic positif
Présentation clinique
Le MU se développe à l’intérieur du globe oculaire et n’est donc pas visible extérieurement. Certains signes indirects existent, provoqués par le décollement partiel de la rétine. Les principaux symptômes sont :
• la baisse d’acuité ou amputation du champ visuel unilatérale,
• un scotome (tache dans le champ de vision),
• des phosphènes intermittents qui se répètent dans le même endroit sur le même œil,
• ou des myodésopsies en cas d’hémorragie du vitré.
On peut aussi observer une dilatation localisée des vaisseaux épiscléraux (3, 11).
Diagnostic
Les MU peuvent être découverts fortuitement lors d’examens ophtalmologiques de routine.
Imagerie
Le fond d’œil est l’examen qui permet le plus souvent le diagnostic. L’ophtalmologue observe une masse tumorale pigmentée (Fig. 1). Le diagnostic est ensuite confirmé par des examens complémentaires tels qu’une angiographie et une échographie de l’œil qui permet de mesurer l’épaisseur de la tumeur (12). L’IRM injectée au gadolinium est conseillée, montrant un hypersignal en T1 et un hyposignal T2. Le scanner thoraco-abdomino-pelvien injecté et l’échographie hépatique permettent de compléter le bilan d’extension initial, à la recherche d’extension locale ou à distance, avant une prise en charge conservatrice. L’histologie n’est pas obligatoire en cas de prise en charge conservatrice par radiothérapie sans chirurgie.

Figure 1 – Fond d’œil de mélanome uvéal par le Dr L. Desjardins (Institut Curie, Paris), photographie issue de la SNOF.
Diagnostics différentiels
Les principaux diagnostics différentiels sont :
• les tumeurs bénignes (hémangiomes, mélanocytomes, ostéomes, nævi)
• et les métastases d’autres cancers, principalement les adénocarcinomes du sein et broncho-
pulmonaires (13).
Pronostic et biologie moléculaire
Classification pronostique
La classification pronostique tient compte :
• des paramètres cliniques du patient (14) et de la tumeur (diamètre basal de la tumeur, épaisseur de la tumeur, atteinte du corps ciliaire, propagation extra-oculaire) (15),
• des paramètres pathologiques (cytomorphologie, néoangiogénèse, nombre élevé de mitoses) (16)
• et des paramètres génétiques.
De nombreux travaux ont mis en évidence le pronostic péjoratif associé à la présence d’une monosomie du chromosome 3 (présence de boucles vasculaires en histologie), mais aussi d’autres altérations chromosomiques telles que : monosomie 1p, trisomie chromosome 8 et altération chromosome 6 (17, 18).
L’identification de groupes pronostiques est essentielle pour sélectionner les patients qui développeront des métastases et qui bénéficieraient d’une intensification du suivi ou des traitements.
Principales mutations
Les principales mutations communes des mélanomes cutanés (BRAF, NRAS, PTEN, TP53 et CDKN2A) sont très peu observées dans les MU primaires (19). Il existe dans plus de 80 % des cas des mutations oncogènes dans les gènes associés aux sous-unités G-protéine-α, Gαq/11, rarement observés dans les primitifs cutanés. Par ailleurs, initialement décrites dans les mélanomes cutanés, les mutations inactivatrices de BAP1 sont trouvées dans 50 % des cas de MU, fréquemment associées à un mauvais pronostic (20). BAP1 code pour la sous-unité catalytique d’une ubiquitine carboxy-terminale hydrolase nucléaire, avec divers substrats, dont BRCA1 et l’histone H2A. Cela conduit à une augmentation du potentiel métastatique du MU, sans que le mécanisme d’action exact soit entièrement élucidé à ce jour (21).
À l’opposé, des mutations associées à un comportement moins agressif, telles que SF3B1 (sous-unité 1 du facteur d’épissage 3B) et EIF1AX (facteur 1A d’initiation à la traduction), sont retrouvées respectivement dans 19 et 24 % des MU (22).
Pistes de recherche
Aucune application clinique n’a découlé de ces observations à ce jour. Mais des pistes se développent en recherche, avec notamment le traitement LXS196, inhibiteur de la protéine kinase C (PKC), qui agit en aval des protéines Gαq/11, et s’est révélé efficace dans les études précliniques (23) (Fig. 2).
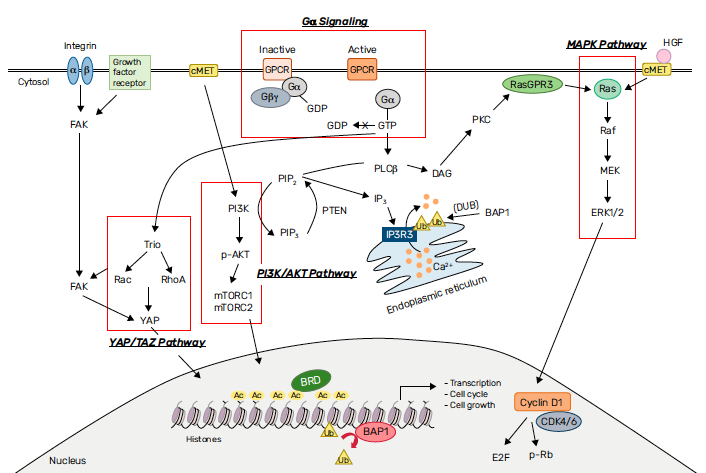
Figure 2 – Voies de signalisation émergentes thérapeutiquement ciblables dans le mélanome uvéal (24).
Traitement
Au stade localisé
L’énucléation et la curiethérapie
L’énucléation est restée pendant longtemps le seul traitement disponible. Une alternative conservatrice par curiethérapie à l’iode 125 s’est développée dans les années 1990 et a démontré des résultats similaires. De nombreuses études rétrospectives et une étude randomisée menée aux États-Unis par le COMS (collaborative ocular melanoma study) n’avaient pas mis en évidence de différence entre les deux traitements (25, 26).
La protonthérapie
L’énucléation a progressivement été supplantée au profit des traitements conservateurs tels que la radiothérapie par protons qui est devenue le traitement standard pour des mélanomes de taille petite à moyenne avec un contrôle local de l’ordre de 95 %. Cette radiothérapie peut être effectuée en France dans l’un des trois centres nationaux dédiés (Institut Curie à Orsay, Centre Lacassagne à Nice et Centre Baclesse à Caen). Elle nécessite au préalable la pose de clip de tantale. Les résultats visuels sont meilleurs lorsque les tumeurs sont petites et ne s’étendent pas à proximité du disque optique et/ou de la fovéa (27). Le risque principal est la baisse de l’acuité visuelle liée au risque de développer une rétinopathie post-radique, un glaucome néovasculaire, une hémorragie intravitréenne ou une neuropathie optique. Une énucléation secondaire peut être alors proposée. L’énucléation primaire reste néanmoins indiquée pour les tumeurs très volumineuses ou localement avancées (extension extra-sclérale, envahissement du nerf optique). Aucun traitement adjuvant n’a montré de bénéfice en termes de survie.
Ces indications de prise en charge à la phase initiale suivent les recommandations spécifiques disponibles à ce jour, établies en 2015 par le Uveal melanoma UK National Guidelines (28).
La surveillance
Au total, plus de 60 % des récidives surviennent dans les 5 premières années et dans 90 % des cas au niveau hépatique. Il est donc proposé aux patients une surveillance avec une échographie hépatique tous les 3 mois pendant 2 ans puis semestrielle pendant 3 ans. L’IRM avec séquences de diffusion peut être préférée chez des patients à haut risque. Il persiste néanmoins un risque de récidive à distance qui peut justifier la poursuite d’une surveillance par échographie au-delà des 5 ans.
Au stade métastatique
En cas de métastases hépatiques uniques
La chirurgie
Il faut systématiquement se poser la question d’une prise en charge chirurgicale carcinologique en cas d’une atteinte limitée accessible à une hépatectomie partielle. Bien qu’il existe peu de données publiées dans la littérature à ce sujet, une étude rétrospective a montré, avec un recul de 16 ans, une médiane de survie de 27 mois en cas de résection R0 (29). Des résultats similaires avaient été rapportés dans une autre étude rétrospective, avec une survie médiane de
25 mois (30).
Les autres traitements locaux
• Des stratégies de traitements intraveineux dirigés directement vers le foie peuvent être proposées comme la chimio-embolisation avec différents agents, dont la fotémustine ou le cisplatine (31).
• Une autre option est la radio-embolisation à l’yttrium-90 (Y-90) récemment évaluée en association avec l’ipilimumab et le nivolumab montrant des signes d’efficacité dans les essais de phases précoces, dont une réponse complète (RC) de 25 mois et deux réponses partielles (RP) de 12 et 16 mois (32). L’étude de phase II SIRTAC, rapportée à l’ASCO 2022, a comparé la radio-embolisation transartérielle Y-90 (SIRT) versus chimio-embolisation trans-artérielle incluant du cisplatine (TACE). La médiane de survie sans progression (mPFS) est de 4,9 versus 2,2 mois respectivement (33).
• Enfin, la perfusion hépatique percutanée (PHP) de Melphalan est une nouvelle alternative permettant de délivrer une forte concentration de chimiothérapie dans le foie. Des données encourageantes avec un taux de réponse objective (ORR) de 47 % ont été publiées dans une analyse rétrospective (34). L’étude de phase III FOCUS a randomisé 144 patients traités soit par Melphalan-PHP toutes les 6-8 semaines, soit par une alternative thérapeutique au choix de l’investigateur (TACE, pembrolizumab, ipilimumab, ou dacarbazine) (35). Les résultats étaient positifs sur l’objectif principal avec un taux de réponse objective de 35,2 % (7 patients en RC et 26 patients en RP) versus 12,5 % (4 patients en RP). La mPFS était de 9,0 versus 3,1 mois. Le profil de toxicité a montré des effets secondaires de grades 3-4 chez 42,6 % des patients traités par Melphalan-PHP, dont la majorité étaient hématologiques, de nature transitoire et résolutive sans séquelles. Aucun décès toxique n’a été observé dans cette étude.
Ainsi, les traitements locaux intrahépatiques semblent induire des réponses tumorales intéressantes et sont des options thérapeutiques à évaluer en monothérapie ou en association avec des immunothérapies.
Une nouvelle thérapie systémique : le tébentafusp
La prise en charge au stade métastatique a été récemment actualisée avec la publication en septembre 2021, par Nathan P. et al, dans The New England of Medicine, d’un essai thérapeutique de phase III, randomisé, multicentrique, en double aveugle, démontrant la supériorité d’une nouvelle thérapeutique, le tébentafusp, comparativement aux traitements standard antérieurs (immunothérapies, chimiothérapies) (36).
Mécanisme d’action
Le tébentafusp (anciennement IMCgp100) est une protéine de fusion bispécifique, appartenant à la catégorie des médicaments immuno-modulateurs. Il redirige les cellules T, à l’aide d’un récepteur soluble de haute affinité pour les lymphocytes T d’un côté et la reconnaissance d’un antigène à la surface des cellules cancéreuses, présenté sous forme de complexe peptide-HLA de l’autre côté (37). Il est spécifique du peptide YLEPGPVTA de la glycoprotéine 100 (gp100), ce fragment intracellulaire d’acides aminés est apprêté et présenté par le HLA-A*02:01 à la surface des cellules de mélanomes uvéaux et cutanés (Fig. 3). L’utilisation du tébentafusp est donc restreinte par l’adressage du peptide cible par le HLA-A*02:01, molécule du complexe majeur d’histocompatibilité humain (CMH-classe I) dont le polymorphisme est exprimé dans environ 45-50 % de la population mondiale.
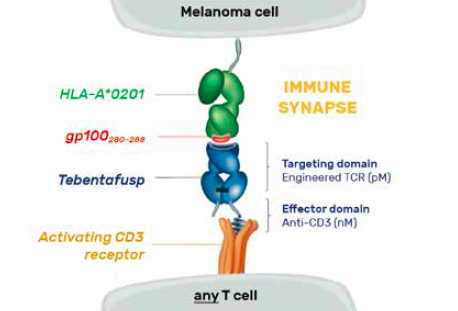
Figure 3 – Mécanisme d’action du tébentafusp (38).
Le mécanisme d’action est de recruter et activer les lymphocytes T polyclonaux du patient, c’est-à-dire indépendamment de leur spécificité TCR, libérant des cytokines et des médiateurs cytolytiques contre les cellules cancéreuses (37, 38).
Essais thérapeutiques
• L’étude de phase I publiée en mars 2022 dans le Journal of clinical oncology avait pour objectif de définir la posologie de ce premier médicament d’une nouvelle classe thérapeutique (protéine bispécifique TCR récepteur/anti-CD3) utilisant un schéma d’escalade hebdomadaire (40). Entre 2016 et 2017, 42 patients ont été inclus dans l’étude et 19 ont reçu une escalade de dose à partir de la troisième semaine selon un design 3+3 pour définir la dose maximale tolérée (DMT) et la dose recommandée pour la phase II (RP2D). Des études pharmacodynamiques et pharmacocinétiques étaient conduites en parallèle. C’est sur la base de cette étude ouverte et internationale qu’a été définie la dose RP2D à 68 μg. Les effets secondaires les plus fréquemment constatés étaient la fièvre (91 %), les rashs cutanés et prurit (83 %), les nausées (74 %), l’asthénie (71 %) et les frissons (69 %). Le traitement était associé à des premiers signes d’efficacité clinique avec un taux de réponse objective d’environ 12 % chez des patients en échec de tout traitement. Des prélèvements sanguins et tumoraux ont permis de montrer que le traitement était associé à une augmentation des cytokines inflammatoires dans le sang et à une majoration de l’infiltrat des lymphocytes T en intra-tumoral.
• Le tébentafusp a ensuite prouvé son efficacité dans une étude clinique de phase II, la survie globale était à 62 % à 12 mois, avec une réduction tumorale de 44 % des lésions cibles chez 116 patients métastatiques prétraités (38).
• Dans l’essai thérapeutique de phase III, 378 patients ont été randomisés (2:1) en première ligne face aux traitements standard au choix de l’investigateur (pembrolizumab, ipilimumab ou dacarbazine) (36). Il a démontré sa supériorité avec une médiane de survie globale (mOS) à 21,7 mois (18,6-28,6) versus 16,0 mois (9,7-18,4) dans le groupe contrôle (HR = 0,51 ; IC 95 % = 0,37-0,71). La mPFS était plus modestement améliorée à 3,3 mois (3,0-5,0) versus 2,9 mois (2,8-3,0), statistiquement significatif. Une analyse supplémentaire a permis d’identifier les patients tirant le plus de bénéfice de ce traitement en première ligne. D’après les données de cette étude, les patients en progression à J100 (indépendamment de leurs critères pronostiques à la baseline) obtenaient une meilleure survie globale lorsqu’ils étaient traités dans le groupe tébentafusp que dans le groupe de traitement standard. Cela était valable également pour les patients dont la maladie était stabilisée à J100, mais l’écart entre les deux groupes était alors plus faible (Fig. 4).
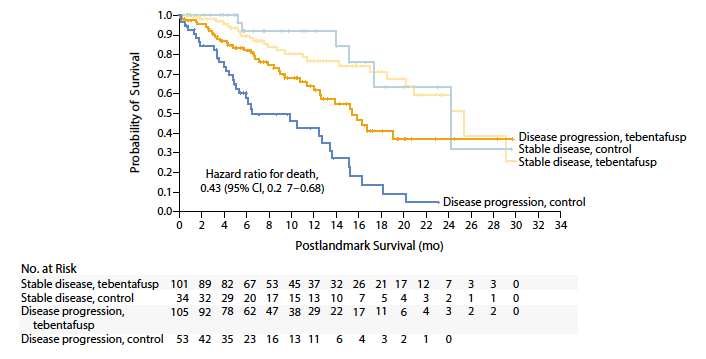
Figure 4 – Survie globale selon la réponse au traitement à J100 (maladie stable ou en progression) (36).
AMM
À la suite de cet essai de phase III, le tébentafusp a fait l’objet le 27 janvier 2022 d’une autorisation d’accès précoce pré-autorisation de mise sur le marché dans l’indication « en monothérapie pour le traitement des patients adultes HLA-A*02 :01-positifs atteints de mélanome uvéal non résécable ou métastatique ». Puis, il a obtenu son AMM dans cette indication, délivrée par l’agence européenne du médicament le 1er avril 2022 sous le nom de Kimmtrak®.
Schéma d’administration
Un cycle comporte 21 jours avec une injection toutes les semaines sans pause. L’introduction du traitement s’effectue avec une escalade de dose, 20 μg à J1, puis 30 μg à J8 et 68 μg à J15. Ces trois premières semaines sont réalisées à l’hôpital avec au minimum une nuit en hospitalisation pour surveiller les réactions systémiques liées au relargage cytokinique. Une fois le palier de 68 μg atteint et supporté, celui-ci est maintenu dans la durée avec une injection par semaine en hôpital de jour. En cas de mauvaise tolérance, il est conseillé de réaliser la cure suivante à la dose du palier inférieur en majorant les traitements prophylactiques (antihistaminiques, antipyrétiques, crème émolliente). Il faut éviter la prise d’une corticothérapie, sauf en cas de symptômes sévères.
Effets secondaires
Les deux principaux effets indésirables liés au tébentafusp résultent des conséquences de la décharge cytokinique libérée par les lymphocytes activés par le traitement d’une part, et par la présence de mélanocytes (positifs à la protéine gp100) dans la peau d’autre part.
Dans l’étude de phase III, 44 % des patients ont rapporté des effets secondaires de grade 3 ou 4, dont 76 % de la fièvre, 83 % des éruptions cutanées et 69 % un prurit (Fig. 5). Ces effets secondaires ont diminué en incidence et en gravité à partir de la quatrième injection et ont rarement conduit à l’arrêt du traitement de l’essai (2 %). Aucun décès lié au traitement n’avait été observé.
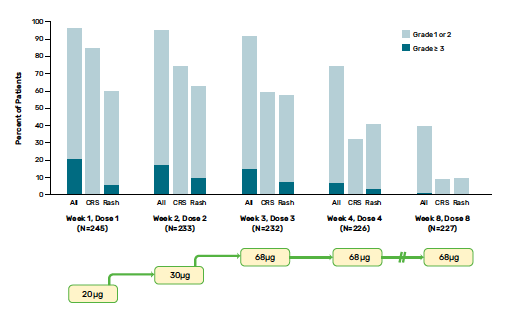
Figure 5 – Incidence et gravité des effets indésirables liés au traitement après les premières doses de tébentafusp (36).
Une grande majorité des patients (89 %) a eu l’apparition d’un syndrome de relargage de cytokines (CRS), selon la définition des critères ASTCT 2019. Il intervenait couramment lors des premières semaines d’injection, dans les 3 à 4 heures suivant l’administration du tébentafusp, le plus souvent avec des symptômes modérés tels que céphalées, nausées, vomissements, fatigue. Il a été rapporté 76 % CRS de grade 2, peu au-delà (1 %). Il est important de graduer la sévérité du CRS pour adapter les traitements (Tab. 1).
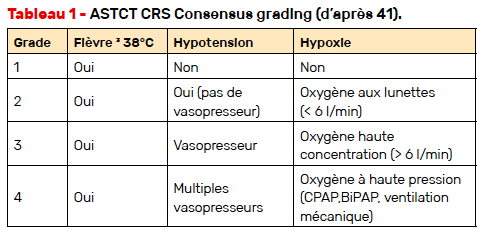
Une vigilance particulière doit être accordée aux paramètres hémodynamiques, car 38 % des patients ont eu des hypotensions post-tébentafusp dans cette étude. Les CRS peuvent également s’accompagner d’œdème facial ou généralisé, et de tremblements.
Le traitement initial des hypotensions est le remplissage vasculaire intraveineux (cristalloïdes ou colloïdes). En cas de persistance d’une hypotension après 2 à 3 heures de remplissage, ou de CRS de grade 3 ou 4, il faut rapidement considérer l’administration de corticostéroïde IV ou tocilizumab (anti-IL-6).
Ainsi, il est recommandé que l’initiation du traitement se réalise dans un centre expert, en hospitalisation courte, habitué à la gestion de ce type d’effets secondaires. Il peut, en second temps, être poursuivi en ambulatoire dans le centre expert ou en relai dans des établissements de soins à proximité du domicile du patient.
Combinaison nivolumab/ipilimumab et positionnement dans les lignes
En monothérapie
Contrairement à la révolution observée par les immune checkpoints inhibiteurs dans la prise en charge des mélanomes cutanés, ces traitements en monothérapie ont montré de très faibles taux de réponse dans le mélanome uvéal métastatique (MUM).
• Un essai de phase II avec ipilimumab en monothérapie avait constaté un ORR de 0 % et une mOS de 6,8 mois (42).
• De même, les anti-PD-1 en monothérapie (nivolumab et pembrolizumab) ont observé des résultats plutôt décevants avec un ORR de 3,6 % et une mOS de 7,6 mois (43).
L’association nivolumab + ipilimumab
Plus récemment, des résultats encourageants ont été rapportés avec l’association des immune checkpoints inhibiteurs.
• Le premier essai de phase II avec la combinaison nivolumab + ipilimumab portait sur 33 patients, dont 43 % avaient reçu une ligne antérieure de traitement (44). Le ORR (critère de jugement principal) était à 18 %, les critères secondaires, mPFS et mOS, étaient à 5,5 et à 19,1 mois respectivement.
• Cette preuve d’efficacité a été confortée par une deuxième étude de phase II (GEM-1402), dans laquelle 52 patients atteints d’un MUM non préalablement traités et non éligibles à une résection hépatique ont reçu du nivolumab + ipilimumab (quatre cycles puis maintenance nivolumab seul) (45). L’objectif principal de l’étude était le taux de survie globale à 12 mois, qui a été évalué à 51,9 %. Parmi les objectifs secondaires, la mOS et la mPFS étaient de 12,7 et de 3,0 mois respectivement. L’ORR était de 51,9 %. L’évaluation de la tolérance a montré un taux important d’effets secondaires sérieux associés au traitement (TR-SAE) chez 30/
52 patients (57,7 %) et 21/52 patients (40,4 %) avec des TR-SAE de grade 3 à 5. Les toxicités graves les plus fréquemment constatées étaient diarrhées (5,8 %), hépatotoxicité (5,8 %), hépatite (3,8 %), thyroïdite (3,8 %) et syndrome de Guillain-Barré (3,8 %).
Ainsi, la combinaison nivolumab + ipilimumab semble également être une piste prometteuse dans la prise en charge du MUM, et pourrait être susceptible de se positionner à l’avenir en concurrence avec le tébentafusp pour la première ligne de traitement.
La séquence tébentafusp et immunothérapie
À l’ESMO 2022 ont été présentés les résultats préliminaires d’une cohorte rétrospective menée sur 20 patients, s’intéressant à la stratégie de séquence du tébentafusp et de l’immunothérapie anti-PD-1 ± anti-CTLA4 (46). Dans cette étude exploratoire, 10 patients étaient traités selon la séquence tébentafusp en L1 puis anti-PD-1 ± anti-CTLA4 en L2 (groupe T-ICI), et 10 patients ont reçu la séquence inverse (groupe ICI-T). Sous réverse de la faible puissance statistique liée à un petit effectif de patients, la PFS et l’OS semblaient allongées dans le groupe T-ICI (mPFS = 2,86 versus 2,35 mois ; HR = 0,38 ; 0,15-1,0) et mOS = 21,47 versus 16,88 mois, non significatif). Une plus large cohorte de validation est nécessaire pour confirmer ces données.
La chimiothérapie
Les molécules utilisées sont la dacarbazine et la fotémustine, avec des taux de réponse très faibles et reléguées en deuxième ou troisième ligne depuis l’arrivée du tébentafusp et de la combinaison anti-PD-1/anti-CTLA4. Étant donné le manque d’efficacité des agents de chimiothérapie, une prise en charge palliative avec des soins de support adaptés sont à considérer en priorité afin de juger au mieux la balance bénéfice/risque de ces traitements.
Conclusion
Le mélanome uvéal est une maladie rare, qui met en jeu le pronostic visuel de l’œil atteint et le pronostic vital. La moitié des patients a une évolution métastatique avec, dans 90 % des cas, des lésions secondaires hépatiques. Le risque, à ce stade, est de développer une insuffisance hépatique sévère, rapidement fatale. Les traitements habituellement efficaces sur le mélanome cutané, comme les anti-PD-1, ne fonctionnent pas ou peu dans le mélanome uvéal. Des études de phase II de combinaison nivolumab + ipilimumab commencent à être rapportées, mais les données en survie semblent encore modestes avec de faibles effectifs de patients. Il n’existe à ce jour aucun traitement médical standard en phase métastatique en dehors du tébentafusp chez les patients HLA-A*02:01, ayant démontré un avantage en survie globale et un contrôle de la charge tumorale. Une vigilance particulière est requise au moment de l’introduction de cette nouvelle thérapie immuno-modulatrice, qui provoque des effets secondaires spécifiques très fréquents et parfois sévères, nécessitant une prise en charge dans des centres expérimentés.
Sylvie Négrier déclare avoir des liens d’intérêt avec Pfizer, Ipsen, BMS, MDS, Eisaï ; Ève-Marie Neidhardt avec Pierre Fabre et Novartis. Mylène Wespiser et Hemerson Guevara déclarent ne pas avoir de liens d’intérêt.
Bibliographie
1. Jager MJ, Shields CL, Cebulla CM et al. Uveal melanoma. Nat Rev Dis Primer 2020 ; 6 : 24.
2. van der Kooij MK, Speetjens FM, van der Burg SH, Kapiteijn E. Uveal versus cutaneous melanoma; same origin, very distinct tumor types. Cancers 2019 ; 11 : E845.
3. Bell DJ, Wilson MW. Choroidal melanoma: natural history and management options. Cancer Control 2004 ; 11 : 296‑303.
4. Vidal JL, Bacin F, Albuisson E et al. [«Melanoma 92». epidemiological study of uveal melanoma in France]. J Fr Ophtalmol 1995 ; 18 : 520‑8.
5. Rantala ES, Hernberg M, Kivelä TT. Overall survival after treatment for metastatic uveal melanoma: a systematic review and meta-analysis. Melanoma Res 2019 ; 29 : 561‑8.
6. Bedikian AY, Legha SS, Mavligit G et al. Treatment of uveal melanoma metastatic to the liver. A review of the M. D. Anderson cancer center experience and prognostic factors. Cancer 1995 ; 76 : 1665‑70.
7. Aronow ME, Topham AK, Singh AD. Uveal melanoma: 5-year update on incidence, treatment, and survival (SEER 1973-2013). Ocul Oncol Pathol 2018 ; 4 : 145‑51.
8. Singh AD, Rennie IG, Seregard S et al. Sunlight exposure and pathogenesis of uveal melanoma. Surv Ophthalmol 2004 ; 49 : 419‑28.
9. Harbour JW, Brantley MA, Hollingsworth H, Gordon M. Association between choroidal pigmentation and posterior uveal melanoma in a white population. Br J Ophthalmol 2004 ; 88 : 39‑43.
10. Klebe S, Driml J, Nasu M et al. BAP1 hereditary cancer predisposition syndrome: a case report and review of literature. Biomark Res 2015 ; 3 : 14.
11. Singh AD, Turell ME, Topham AK. Uveal melanoma: trends in incidence, treatment, and survival. Ophthalmology 2011 ; 118 : 1881‑5.
12. Coleman DJ, Silverman RH, Rondeau MJ et al. Noninvasive in vivo detection of prognostic indicators for high-risk uveal melanoma: Ultrasound parameter imaging. Ophthalmology 2004 ; 111 : 558‑64.
13. Jardel P, Sauerwein W, Olivier T et al. Management of choroidal metastases. Cancer Treat Rev 2014 ; 40 : 1119‑28.
14. Lorenzo D, Piulats JM, Ochoa M et al. Clinical predictors of survival in metastatic uveal melanoma. Jpn J Ophthalmol 2019 ; 63 : 197‑209.
15. Schmittel A, Bechrakis NE, Martus P et al. Independent prognostic factors for distant metastases and survival in patients with primary uveal melanoma. Eur J Cancer 2004 ; 40 : 2389‑95.
16. McLean IW, Keefe KS, Burnier MN. Uveal melanoma: comparison of the prognostic value of fibrovascular loops, mean of the ten largest nucleoli, cell type, and tumor size. Ophthalmology 1997 ; 104 : 777‑80.
17. Sisley K, Parsons MA, Garnham J et al. Association of specific chromosome alterations with tumour phenotype in posterior uveal melanoma. Br J Cancer 2000 ; 82 : 330‑8.
18. Bornfeld N, Prescher G, Becher R et al. Prognostic implications of monosomy 3 in uveal melanoma. Lancet 1996 ; 347 : 1222‑5.
19. Edmunds SC, Cree IA, Dí Nícolantonío F et al. Absence of BRAF gene mutations in uveal melanomas in contrast to cutaneous melanomas. Br J Cancer 2003 ; 88 : 1403‑5.
20. Sun M, Zhou W, Qi X et al. Prediction of BAP1 expression in uveal melanoma using densely-connected deep classification networks. Cancers 2019 ; 11 : 1579.
21. Koopmans AE, Verdijk RM, Brouwer RWW et al. Clinical significance of immunohistochemistry for detection of BAP1 mutations in uveal melanoma. Mod Pathol 2014 ; 27 : 1321‑30.
22. Martin M, Maßhöfer L, Temming P et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet 2013 ; 45 : 933‑6.
23. Chua V, Lapadula D, Randolph C et al. Dysregulated GPCR signaling and therapeutic options in uveal melanoma. Mol Cancer Res 2017 ; 15 : 501‑6.
24. Chua V, Mattei J, Han A et al. The latest on uveal melanoma research and clinical trials: updates from the Cure Ocular Melanoma (CURE OM) Science Meeting (2019). Clin Cancer Res 2021 ; 27 : 28-33.
25. Puusaari I, Heikkonen J, Summanen P et al. Iodine brachytherapy as an alternative to enucleation for large uveal melanomas. Ophthalmology 2003 ; 110 : 2223‑34.
26. Hawkins BS. Collaborative ocular melanoma study randomized trial of I-125 brachytherapy. Clin Trials Lond Engl 2011 ; 8 : 661‑73.
27. Dendale R, Rouic LLL, Noel G et al. Proton beam radiotherapy for uveal melanoma: Results of Curie Institut-Orsay Proton Therapy Center (ICPO). Int J Radiat Oncol Biol Phys 2006 ; 65 : 780‑7.
28. Nathan P, Cohen V, Coupland S et al. Uveal melanoma UK national guidelines. Eur J Cancer 2015 ; 51 : 2404-12.
29. Mariani P, Piperno-Neumann S, Servois V et al. Surgical management of liver metastases from uveal melanoma: 16 years’ experience at the Institut Curie. Eur J Surg Oncol 2009 ; 35 : 1192‑7.
30. Rivoire M, Kodjikian L, Baldo S et al. Treatment of liver metastases from uveal melanoma. Ann Surg Oncol 2005 ; 12 : 422‑8.
31. Schuster R, Lindner M, Wacker F et al. Transarterial chemoembolization of liver metastases from uveal melanoma after failure of systemic therapy: toxicity and outcome. Melanoma Res 2010 ; 20 : 191‑6.
32. Levey AO, Elsayed M, Lawson DH et al. Predictors of overall and progression-free survival in patients with ocular melanoma metastatic to the liver undergoing Y90 radioembolization. Cardiovasc Intervent Radiol 2020 ; 43 : 254‑63.
33. Peuker CA, De Bucourt M, Gebauer B et al. First interim analysis of the SirTac trial: A randomized phase II study of SIRT and DSM-TACE in patients with liver metastases from uveal melanoma. J Clin Oncol 2022 ; 40 : 9511.
34. Karydis I, Gangi A, Wheater MJ et al. Percutaneous hepatic perfusion with melphalan in uveal melanoma: A safe and effective treatment modality in an orphan disease. J Surg Oncol 2018 ; 117 : 1170‑8.
35. Zager JS, Orloff MM, Ferrucci PF et al. FOCUS phase 3 trial results: Percutaneous hepatic perfusion (PHP) with melphalan for patients with ocular melanoma liver metastases (PHP-OCM-301/301A). J Clin Oncol 2022 ; 40 : 9510.
36. Nathan P, Hassel JC, Rutkowski P et al. Overall survival benefit with tebentafusp in metastatic uveal melanoma. N Engl J Med 2021 ; 385 : 1196‑206.
37. Boudousquie C, Bossi G, Hurst JM et al. Polyfunctional response by ImmTAC (IMCgp100) redirected CD8+ and CD4+ T cells. Immunology 2017 ; 152 : 425‑38.
38. Sacco J, Carvajal R, Butler MO et al. A phase (ph) II, multi-center study of the safety and efficacy of tebentafusp (tebe) (IMCgp100) in patients (pts) with metastatic uveal melanoma (mUM). Ann Oncol 2022 ; 31 : 64MO.
39. Middleton MR, McAlpine C, Woodcock VK et al. Tebentafusp, A TCR/Anti-CD3 bispecific fusion protein targeting gp100, potently activated antitumor immune responses in patients with metastatic melanoma. Clin Cancer Res 2020 ; 26 : 5869‑78.
40. Carvajal RD, Nathan P, Sacco JJ et al. Phase I study of safety, tolerability, and efficacy of tebentafusp using a step-up dosing regimen and expansion in patients with metastatic uveal melanoma. J Clin Oncol 2022 ; 40 : 1939‑48.
41. Lee DW, Santomasso BD, Locke FL et al. ASTCT Consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells. Biol Blood Marrow Transplant 2019 ; 25 : 625‑38.
42. Zimmer L, Vaubel J, Mohr P et al. Phase II DeCOG-study of ipilimumab in pretreated and treatment-naïve patients with metastatic uveal melanoma. PLOS ONE 2015 ; 10 : e0118564.
43. Algazi AP, Tsai KK, Shoushtari AN et al. Clinical outcomes in metastatic uveal melanoma treated with PD-1 and PD-L1 antibodies: PD-1 blockade in uveal melanoma. Cancer 2016 ; 122 : 3344‑53.
44. Pelster MS, Gruschkus SK, Bassett R et al. Nivolumab and ipilimumab in metastatic uveal melanoma: results from a single-arm phase II study. J Clin Oncol 2021 ; 39 : 599‑607.
45. Piulats JM, Espinosa E, de la Cruz Merino L et al. Nivolumab plus ipilimumab for treatment-naïve metastatic uveal melanoma: an open-label, multicenter, phase II trial by the spanish multidisciplinary melanoma group (GEM-1402). J Clin Oncol 2021 ; 39 : 586‑98.
46. Koch EC, Arteag DP, Mantle L et al. Outcomes of immune checkpoint. Ann Oncol 2022 ; 33 : 831P.